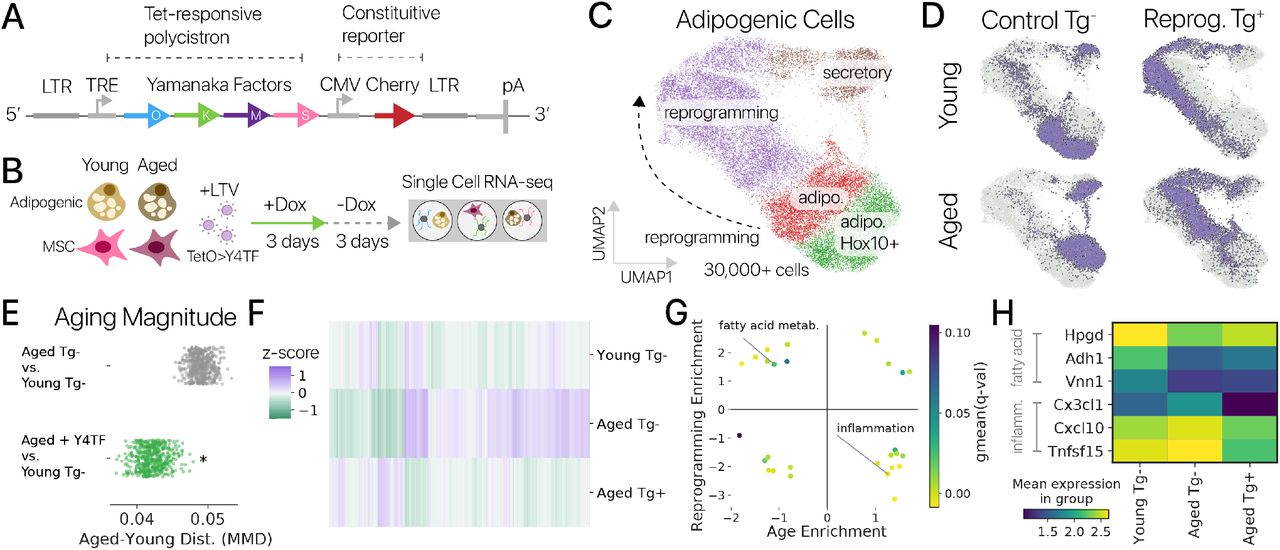
Авторы: Александр Трапп, Чаба Керепеси, Вадим Николаевич Гладышев
Введение
Один из канонических признаков старения это глубокие эпигенетические изменения, особенно в динуклеотидах CG (CpG) ( Horvath & Raj, 2018 ; López-Otín et al., 2013 ). Изменения в метилировании CpG с возрастом теперь можно анализировать с использованием различных подходов, от массивов гибридизации до полногеномных или целевых методов секвенирования следующего поколения ( Han, Franzen, et al., 2020 ; Horvath, 2013 ; Lister et al. , 2009 ; Meissner et al., 2005 ). Эти методы позволяют проводить количественное исследование динамического ландшафта метилирования ДНК с точностью до одного нуклеотида в любой интересующей ткани организмов, развивших данный тип регуляции, таких как млекопитающие.
С момента своего создания в последнее десятилетие прогнозирующие многовариантные модели машинного обучения, основанные на уровнях метилирования ДНК (DNAm), получившие название `` эпигенетические часы '', произвели революцию в области изучения старения ( Bocklandt et al., 2011 ; Horvath, 2013 ). Часы, изначально построенные лишь для оценки хронологического возраста, теперь могут также интегрировать и прогнозировать различные показатели биологического старения и риска заболеваний, что подчеркивает их клиническую значимость ( Levine et al., 2018 ; Lu et al., 2019 ). Интересно, что недавно было разработано несколько пан-тканевых часов для млекопитающих, которые могут с впечатляющей точностью определять эпигенетический возраст практически в любой ткани млекопитающих ( Mammalian Methylation Consortium, 2021). Помимо этих замечательных достижений, эпигенетические часы представляют особый интерес, поскольку эти модели обещают обнаруживать даже небольшие изменения биологического возраста в результате различных интервенций для увеличения продолжительности жизни или перепрограммирования клеток ( Lu et al., 2020 ; Petkovich и др., 2017 ).
Однако, хотя единицами жизни являются отдельные клетки, все существующие эпигенетические часы полагаются на измерения, полученные из массивных образцов (то есть образцов, содержащих множество клеток), как для создания, так и для применения этих моделей ( Bell et al., 2019 ). Исторически сложилось так, что использование массивных образцов для анализа метилирования ДНК было неотъемлемым требованием доступных методологий, которые требовали сотни нанограмм исходного материала из-за жесткой химической обработки ДНК бисульфитом ( Karemaker & Vermeulen, 2018 ). Хотя использование объемной ткани и удобно, оно по сути скрывает эпигенетическую гетерогенность, существующую между отдельными клетками ( Bell et al., 2019 ; Gravina et al., 2016). Есть недавняя работа , где были рассмотрены характеристики транскриптомных изменений при старении мышей на уровне отдельных клеток, но детальные эпигенетические изменения отдельных клеток и тканей во время старения млекопитающих остаются в основном неизученными ( Almanzar et al., 2020 ).
Недавние успехи в методах эпигеномного секвенирования позволили оценить ограниченные профили метилирования в отдельных клетках. С момента появления этих методов за предыдущее десятилетие появилось множество методов секвенирования метилирования отдельных клеток, в том числе бисульфитное секвенирование с пониженным представлением отдельных клеток
(scRRBS) и бисульфитное секвенирование одной клетки (весь геном) (scWGBS / scBS) ( Gravina et al. al., 2016 ; Guo et al., 2013 ; Smallwood et al., 2014). Эти подходы основаны на ключевых модификациях оригинальных методик с объемными образцами, которые позволяют снизить потерю ДНК во время подготовки библиотеки. Интересно, что были также разработаны новые методы секвенирования РНК, метилирования ДНК и доступности хроматина в одной и той же клетке, что позволяет интегрировать мультиомиксные анализы с максимальным разрешением ( Angermueller et al., 2016 ; Argelaguet et al., 2019 ; Кларк и др., 2018 ).
Несмотря на этот замечательный прогресс в омиках одиночных клеток, остаются общие проблемы разреженности. В зависимости от конкретного используемого метода, только небольшая часть CpG, охваченных методами массового секвенирования, представлена в отдельных клетках ( рис. 1а , рис. S1 ). Более того, наиболее распространенные протоколы для профилирования метиломов отдельных клеток - те, которые включают в себя исследование паттернов метилирования ДНК в масштабе всего генома - дополнительно страдают от эффективного случайного охвата считываний ( Karemaker & Vermeulen, 2018 ). Чтобы преодолеть этот ограничивающий фактор, анализ профилей метилирования отдельных клеток обычно проводится путем усреднения уровней метилирования в геномных областях ( Angermueller et al., 2016 ; Luo et al., 2017). В качестве альтернативы также было разработано несколько стратегий вменения и кластеризации, использующих байесовские подходы или подходы глубокого обучения для заполнения недостающих состояний метилирования для CpG, не охваченных в данной клетке ( Angermueller et al., 2017 ; Kapourani & Sanguinetti, 2019 ). Хотя эти методы вменения работают очень хорошо при различении клеточных подтипов друг от друга, они полагаются на построение моделей для конкретных наборов данных, что затрудняет проведение объективных сравнений между исследованиями.
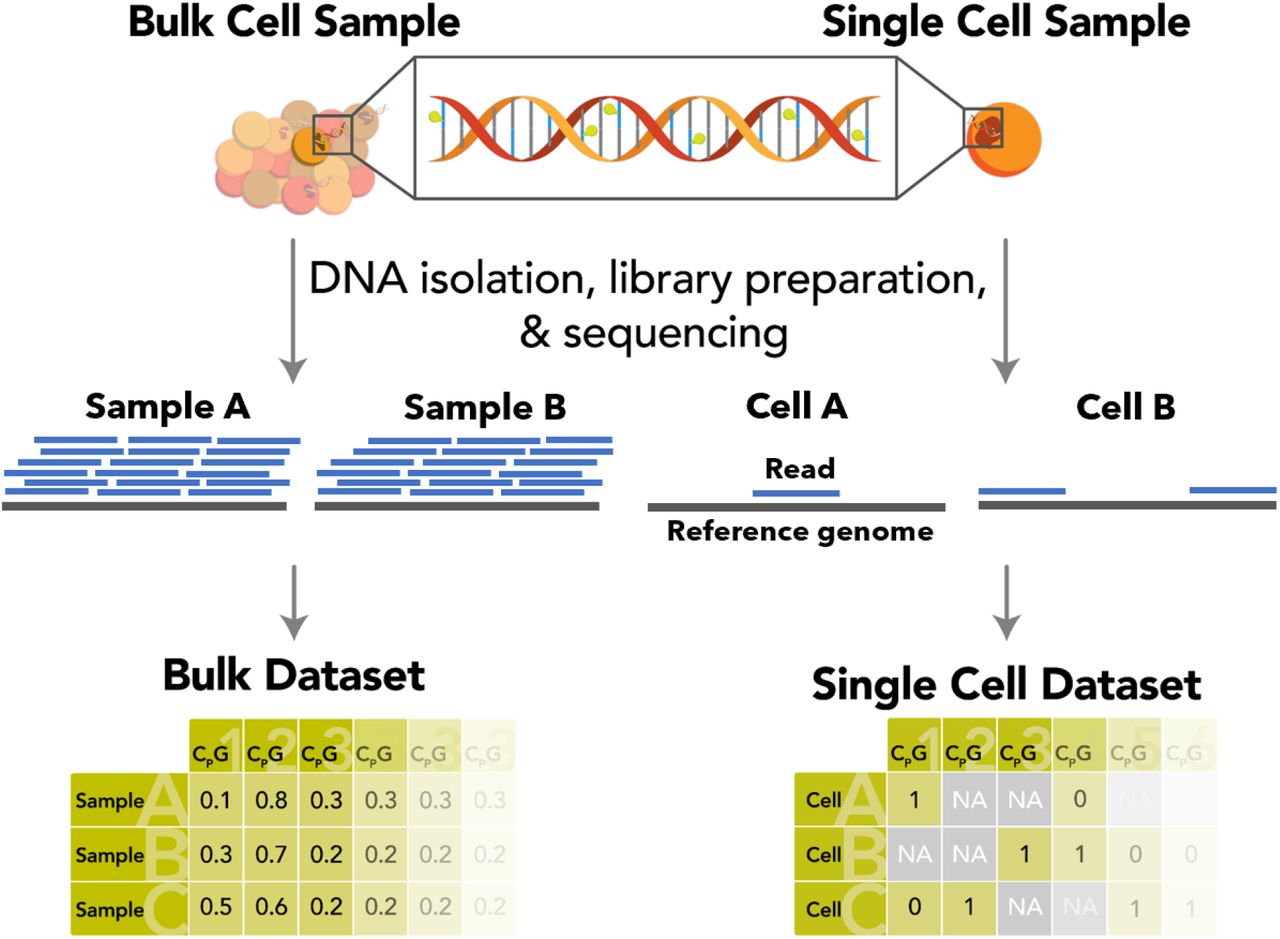 Рисунок S1: Практические различия в результатах методов массового и одноклеточного эпигенетического секвенирования Схематическое сравнение подходов к секвенированию массового (слева) и одноклеточного (справа) метилирования. Использование объемных выборок обеспечивает высокий и постоянный охват CpG, в то время как профили отдельных клеток обычно страдают от эффективного случайного и разреженного покрытия. Объемные образцы дают непрерывные значения от 0 до 1 (из-за наличия считываний из разных клеток), в то время как образцы отдельных клеток демонстрируют в основном дигитальное метилирование. Следовательно, обычные таблицы характеристик (внизу), используемые для обучения часов регрессии эластичной сети, невозможно создать для отдельных клеток из-за наличия большого количества недостающих данных.
Рисунок S1: Практические различия в результатах методов массового и одноклеточного эпигенетического секвенирования Схематическое сравнение подходов к секвенированию массового (слева) и одноклеточного (справа) метилирования. Использование объемных выборок обеспечивает высокий и постоянный охват CpG, в то время как профили отдельных клеток обычно страдают от эффективного случайного и разреженного покрытия. Объемные образцы дают непрерывные значения от 0 до 1 (из-за наличия считываний из разных клеток), в то время как образцы отдельных клеток демонстрируют в основном дигитальное метилирование. Следовательно, обычные таблицы характеристик (внизу), используемые для обучения часов регрессии эластичной сети, невозможно создать для отдельных клеток из-за наличия большого количества недостающих данных.
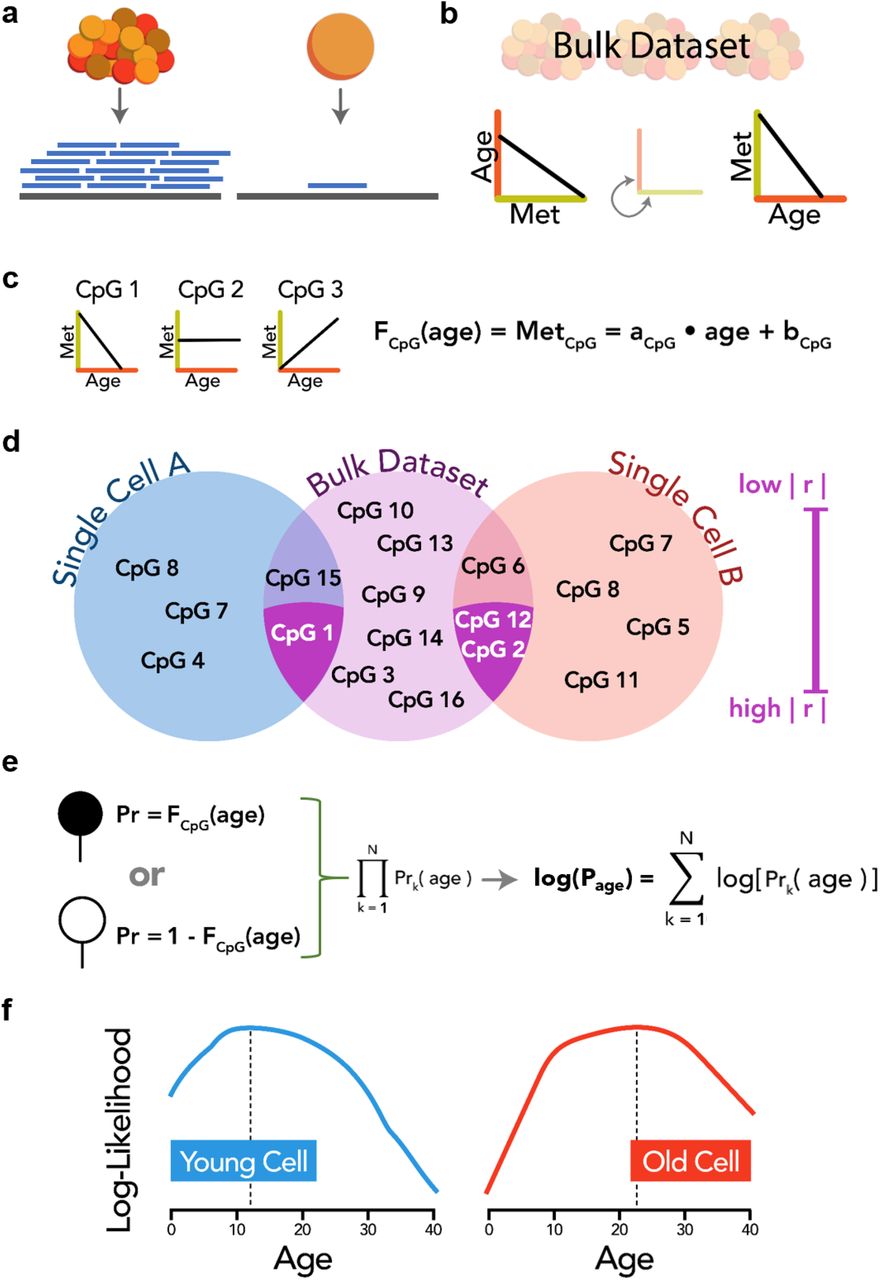 Рисунок 1: Разработка scAge а) Схема дифференциального выравнивания считывания с помощью с группового (слева) и отдельноклеточного (справа) методов эпигеномного секвенирования. Уменьшение ввода ДНК для отдельных клеток приводит к меньшему количеству отображенных считываний (синий) в эталонном геноме (серый), что приводит к уменьшению покрытия CpG по сравнению с объемными образцами. б) Схематическое изменение линейной зависимости между уровнем метилирования и возрастом. Используя групповые образцы разного возраста, можно отобразить, как уровень метилирования (зеленая ось) влияет или предсказывает возраст (красная ось). Напротив, можно установить и обратное соотношение, при котором возраст может использоваться в качестве предиктора среднего уровня метилирования. в) Вычисление формул линейной регрессии для каждого CpG в наборе обучающих данных. Уровень метилирования CpG может увеличиваться, уменьшаться или не иметь явной линейной зависимости с возрастом. Met обозначает средний уровень метилирования, a указывает наклон линейного уравнения, а b представляет собой точку пересечения вдоль оси метилирования. г) Схематическое пересечение и этап фильтрации scAge. CpG в наборе обучающих данных (розовый) пересекаются с таковыми в отдельных клетках (синий / оранжевый). CpG в одной отдельной клетке показывают ограниченные пересечения с CpG в другой отдельной клетке. Общие CpG между любой одной клеткой и набором данных массового обучения ранжируются на основе их абсолютной корреляции | r | с возрастом. Только верхние коррелированные общие CpG (темно-фиолетовые пересечения) в каждой клетке принимаются в качестве входных данных для алгоритма scAge. д) Схема основного алгоритма scAge. Апостериорные вероятности вычисляются на основе наблюдаемых состояний метилирования в отдельных клетках для каждого возрастного шага. Закрашенный кружок указывает на метилированный CpG, а белый кружок указывает на неметилированный CpG в конкретной отдельной клетке. Метрика правдоподобия для профиля вычисляется как произведение всех независимых вероятностей на CpG. На практике это оценивается как сумма логарифма правдоподобия для преодоления вычислительных ограничений потери значимости. е) Схематическое представление распределения зависящих от возраста показателей логарифмического правдоподобия в молодой и старой клетках.
Рисунок 1: Разработка scAge а) Схема дифференциального выравнивания считывания с помощью с группового (слева) и отдельноклеточного (справа) методов эпигеномного секвенирования. Уменьшение ввода ДНК для отдельных клеток приводит к меньшему количеству отображенных считываний (синий) в эталонном геноме (серый), что приводит к уменьшению покрытия CpG по сравнению с объемными образцами. б) Схематическое изменение линейной зависимости между уровнем метилирования и возрастом. Используя групповые образцы разного возраста, можно отобразить, как уровень метилирования (зеленая ось) влияет или предсказывает возраст (красная ось). Напротив, можно установить и обратное соотношение, при котором возраст может использоваться в качестве предиктора среднего уровня метилирования. в) Вычисление формул линейной регрессии для каждого CpG в наборе обучающих данных. Уровень метилирования CpG может увеличиваться, уменьшаться или не иметь явной линейной зависимости с возрастом. Met обозначает средний уровень метилирования, a указывает наклон линейного уравнения, а b представляет собой точку пересечения вдоль оси метилирования. г) Схематическое пересечение и этап фильтрации scAge. CpG в наборе обучающих данных (розовый) пересекаются с таковыми в отдельных клетках (синий / оранжевый). CpG в одной отдельной клетке показывают ограниченные пересечения с CpG в другой отдельной клетке. Общие CpG между любой одной клеткой и набором данных массового обучения ранжируются на основе их абсолютной корреляции | r | с возрастом. Только верхние коррелированные общие CpG (темно-фиолетовые пересечения) в каждой клетке принимаются в качестве входных данных для алгоритма scAge. д) Схема основного алгоритма scAge. Апостериорные вероятности вычисляются на основе наблюдаемых состояний метилирования в отдельных клетках для каждого возрастного шага. Закрашенный кружок указывает на метилированный CpG, а белый кружок указывает на неметилированный CpG в конкретной отдельной клетке. Метрика правдоподобия для профиля вычисляется как произведение всех независимых вероятностей на CpG. На практике это оценивается как сумма логарифма правдоподобия для преодоления вычислительных ограничений потери значимости. е) Схематическое представление распределения зависящих от возраста показателей логарифмического правдоподобия в молодой и старой клетках.
Общая разреженность профилей DNAm отдельных клеток создает серьезные ограничения для создания отдельноклеточных эпигенетических часов. Построение этих прогностических моделей традиционно основывалось на сборе уровней метилирования CpG, которые последовательно охватывались между выборками разного возраста ( Meer et al., 2018 ; Stubbs et al., 2017 ; Thompson et al., 2018 ). В объемной ткани это позволяет создавать большие таблицы характеристик, которые затем можно напрямую использовать для машинного обучения, в частности, для эластичной сетевой регрессии ( Zou & Hastie, 2005 ). Однако разреженные и бинаризованные профили метилирования отдельных клеток препятствуют применению этого подхода ( рис. S1 ) ( Bell et al., 2019). Несмотря на эти проблемы, создание отдельноклеточных эпигенетических часов обещает новые методы эпигенетического профилирования возраста со сверхнизким входом в сочетании с беспрецедентно подробным наблюдением процесса старения.
Здесь мы разработали scAge, новый метод, способный определять эпигенетический возраст отдельных клеток. Из-за низкого и непоследовательного покрытия CpG наш подход вместо этого полагается на вероятностный алгоритм, который в значительной степени не зависит от того, какие CpG покрываются в каждой клетке. Используя линейную взаимосвязь уровней метилирования с возрастом в подмножестве CpG, мы создаем показатель правдоподобия, который количественно определяет вероятность того, что клетка происходит из основной выборки данного возраста. Наш метод воспроизводит хронологический возраст ткани в среднем, а также раскрывает внутреннюю эпигенетическую гетерогенность, существующую между клетками. Использование этих вероятностных эпигенетических часов открывает новые захватывающие возможности для исследований биологического старения на ранее неуловимом уровне отдельных клеток.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Разработка scAge: вероятностные отдельноклеточные эпигенетические часы
Основными проблемами при оценке возраста одиночных клеток являются их разреженные и бинаризованные профили метилирования. В отличие от массовых выборок, последовательные считывания охватывают разные части генома каждой отдельной клетки с очень низким перекрытием между клетками ( рис. 1a , рис. S1.). Чтобы преодолеть эти ограничения, мы предположили, что уровни метилирования сайтов CpG с высоким покрытием при групповом секвенировании или профилировании массива DNAm ткани предлагают оценку вероятности метилирования в этих конкретных сайтах CpG в любой отдельной клетке, происходящей из этой ткани. Кроме того, мы перевернули общепринятое представление о взаимосвязи между уровнем метилирования и возрастом: в то время как текущие групповые часы используют уровень метилирования в качестве предиктора возраста, мы предположили, что возраст можно рассматривать как предиктор уровня группового метилирования для любого данного CpG ( рис. . 1b ). Используя данные обучения, полученные из групповых RRBS, мы оценили изменение средних уровней метилирования с возрастом для каждого CpG в этом эталонном наборе, используя одномерные линейные модели ( Fig. 1c ).
Затем мы изолировали общие сайты CpG между любым заданным профилем отдельных клеток и эталонным набором данных вероятности метилирования ( рис. 1d) . Затем мы выбрали определенное количество общих CpG, которые показали наибольшую абсолютную корреляцию Пирсона с возрастом в массиве данных (т. Е. Связанных с возрастом сайтов CpG). Важно отметить, что из-за разреженности профилей DNAm отдельных клеток, покрытые сайты CpG сильно различаются от клетки к клетке; несмотря на это, небольшая, но отчетливая коллекция ассоциированных с возрастом сайтов CpG была охвачена в каждой секвенированной клетке ( Fig. 1d ).
Затем мы вычислили вероятность наблюдения этого профильтрованного профиля метилирования отдельной клетки в любом заданном возрасте ( Рис. 1e ). На практике мы использовали логарифмы (логарифм правдоподобия), чтобы избежать ошибок переполнения во время вычислений. Наконец, мы определили возраст, для которого эта вероятность максимальна ( рис. 1е ). Мы обнаружили, что этот подход, который мы назвали scAge, позволяет точно определить эпигенетический возрастной профиль в отдельных клетках с очень разными и разреженными профилями метиломов.
Вероятностные одноклеточные часы рекапитулируют хронологический возраст отдельных гепатоцитов.
Сначала мы применили scAge к терминально дифференцированным клеткам очень молодых и очень старых мышей, включая 11 одиночных гепатоцитов от 4-месячных животных и 10 одиночных гепатоцитов от 26-месячных животных ( Gravina et al., 2016 ). Профили отдельных клеток содержали ограниченное число общих CpG для любой взятой пары клеток; фактически, этот эффект усиливается, когда сайты в дополнительных клетках прогрессивно пересекаются, что приводит к минимальному окончательному перекрытию ( Fig. 2a ). Охват варьировал от 0,4 до 3,2 миллиона CpG в гепатоцитах со сходным средним глобальным метилированием в молодых и старых клетках ( рис. 2b , рис. S2a ). Сначала мы применили наши вероятностные часы, обученные на объемных образцах печени ( рис. 2c ) ( Thompson et al., 2018). Примечательно, что при использовании только 700 независимых CpG на клетку scAge продемонстрировал впечатляющую точность и согласованность в предсказаниях возраста молодых и старых гепатоцитов ( рис. 2d ). Мы достигли коэффициента Пирсона 0,88 со средней и медианной абсолютными ошибками 3,9 и 2,9 месяца соответственно. Таким образом, scAge печени правильно воспроизводит возраст исходной ткани с помощью небольшого количества клеток.
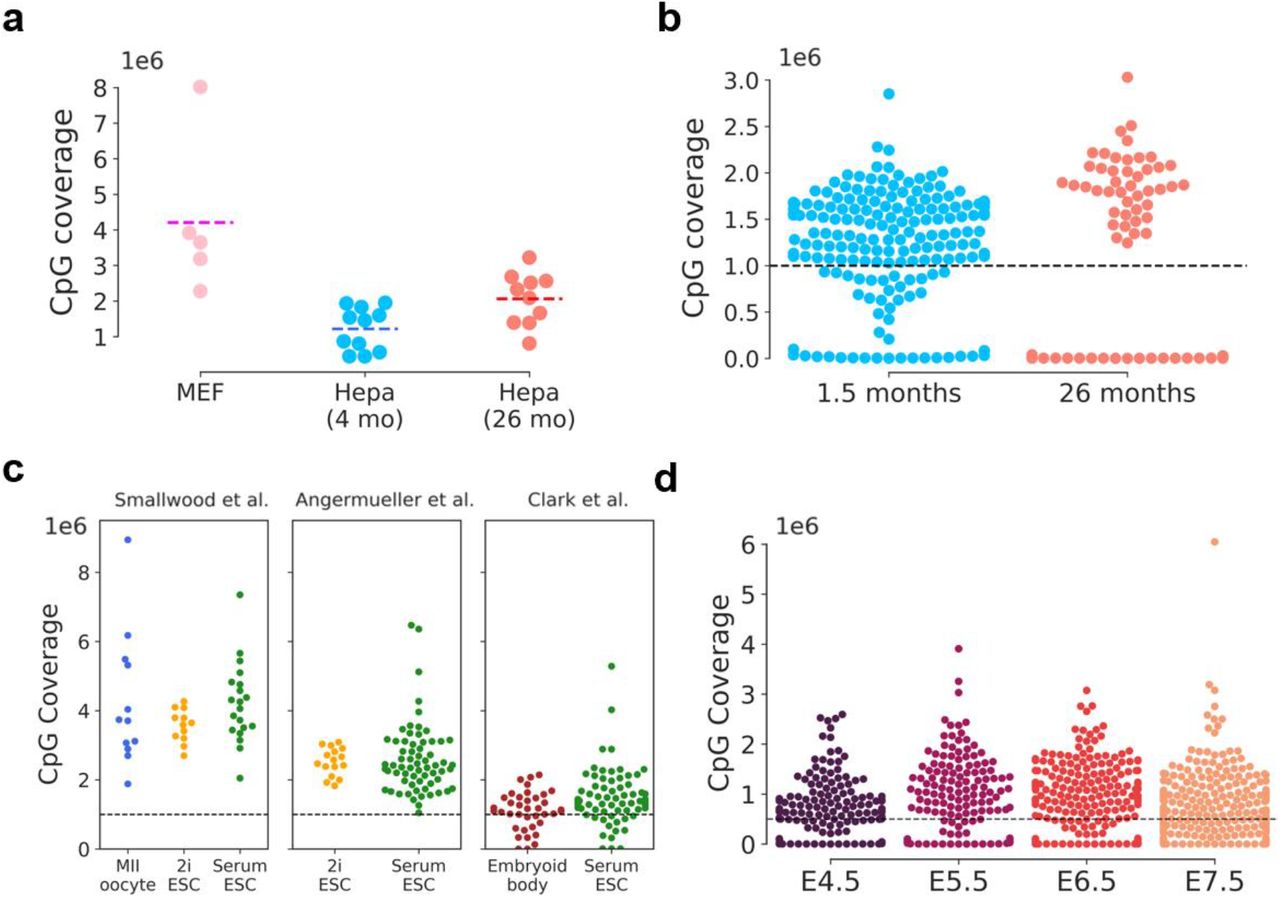 Рисунок S2:Покрытие одиночной клетки CpG варьируется в зависимости от набора данных Охват CpG в (а) MEF, гепатоциты 4- и 26-месячного возраста; (б) 1,5-месячные и 26-месячные мышечные стволовые клетки; (c) ооциты MII, ESC, культивируемые в условиях 2i или сыворотки, и эмбриоидные тельца; и (d) эмбриональные клетки от E4.5 до E7.5. На основе профиля покрытия и количества клеток в каждом наборе данных применялась фильтрация для удаления клеток с очень низким покрытием (черные пунктирные линии). Пороговое значение не было установлено в (a) из-за низкого количества клеток, пороговое значение в 1000000 CpG было обозначено в (b) и (c) , а пороговое значение в 500000 CpG было установлено в (d) для оптимизации как согласованности прогнозов, так и наибольшего количество клеток на исследование.
Рисунок S2:Покрытие одиночной клетки CpG варьируется в зависимости от набора данных Охват CpG в (а) MEF, гепатоциты 4- и 26-месячного возраста; (б) 1,5-месячные и 26-месячные мышечные стволовые клетки; (c) ооциты MII, ESC, культивируемые в условиях 2i или сыворотки, и эмбриоидные тельца; и (d) эмбриональные клетки от E4.5 до E7.5. На основе профиля покрытия и количества клеток в каждом наборе данных применялась фильтрация для удаления клеток с очень низким покрытием (черные пунктирные линии). Пороговое значение не было установлено в (a) из-за низкого количества клеток, пороговое значение в 1000000 CpG было обозначено в (b) и (c) , а пороговое значение в 500000 CpG было установлено в (d) для оптимизации как согласованности прогнозов, так и наибольшего количество клеток на исследование.
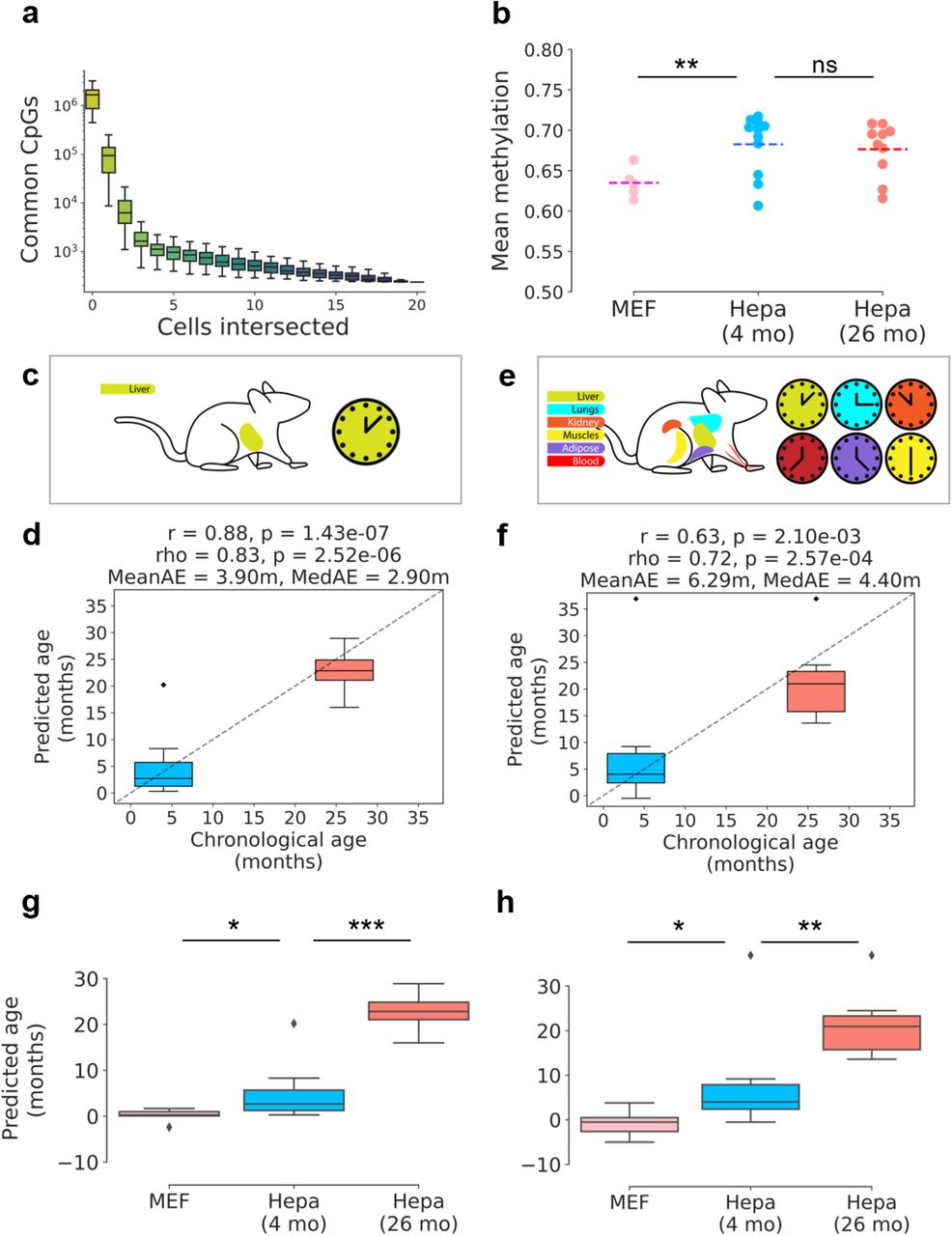 Рисунок 2:scAge рекапитулирует возраст гепатоцитов и эмбриональных фибробластов а) Прогрессивное пересечение сайтов CpG в каждом отдельном гепатоците. Порядок пересечений менялся 100 раз, чтобы получить распределения для каждой добавленной дополнительной клетки. Ось Y логарифмически преобразована. Минимальные общие CpG остаются после пересечения всех клеток. б) Среднее метилирование в MEF, 4-месячных и 26-месячных гепатоцитах. ** означает p <0,01. в) Схематическое изображение часов scAge печени. г) Прогнозирование эпигенетического возраста для всех молодых и старых гепатоцитов с использованием часов scAge печени. д) Схематическое изображение мультиканальных часов. Были включены ткани печени, легких, почек, мышц, жировая ткань и кровь. е) Прогнозирование эпигенетического возраста для всех молодых и старых гепатоцитов с использованием мультитканевых часов SCAge. Применение часов ScAge печени (g) и часов scAge для нескольких тканей (h) на эмбриональных фибробластах мыши (MEF) по сравнению с молодыми и старыми гепатоцитами. * обозначает p <0,05, ** обозначает p <0,01, а *** обозначает p <0,001.
Рисунок 2:scAge рекапитулирует возраст гепатоцитов и эмбриональных фибробластов а) Прогрессивное пересечение сайтов CpG в каждом отдельном гепатоците. Порядок пересечений менялся 100 раз, чтобы получить распределения для каждой добавленной дополнительной клетки. Ось Y логарифмически преобразована. Минимальные общие CpG остаются после пересечения всех клеток. б) Среднее метилирование в MEF, 4-месячных и 26-месячных гепатоцитах. ** означает p <0,01. в) Схематическое изображение часов scAge печени. г) Прогнозирование эпигенетического возраста для всех молодых и старых гепатоцитов с использованием часов scAge печени. д) Схематическое изображение мультиканальных часов. Были включены ткани печени, легких, почек, мышц, жировая ткань и кровь. е) Прогнозирование эпигенетического возраста для всех молодых и старых гепатоцитов с использованием мультитканевых часов SCAge. Применение часов ScAge печени (g) и часов scAge для нескольких тканей (h) на эмбриональных фибробластах мыши (MEF) по сравнению с молодыми и старыми гепатоцитами. * обозначает p <0,05, ** обозначает p <0,01, а *** обозначает p <0,001.
Хотя scAge хорошо помогает интегрировать прогнозы для нескольких отдельных клеток в точный предиктор общего возраста ткани, он также по своей сути обеспечивает повышенное разрешение вплоть до отдельных клеток. Действительно, некоторые клетки в одной и той же ткани были моложе или старше других. Самый низкий прогноз с использованием печеночных часов для молодых клеток был близок к 0, при этом возраст одной клетки был около 20 месяцев. Эти результаты предполагают глубокую гетерогенность процесса старения, при котором глобальные изменения эпигенетического возраста в основной ткани характеризуются неравномерными и разнообразными траекториями старения, которым подвергаются отдельные клетки.
Мы также использовали scAge, обученный на наборах данных по тканям почек, крови, печени, легких, мышц и жировой ткани ( рис. 2e ) ( Thompson et al., 2018 ). Поскольку мультитканевые наборы данных накладывают биологический шум на взаимосвязь между возрастом и уровнем метилирования в большинстве CpG, абсолютные корреляции между обеими переменными резко упали по сравнению с набором данных только для печени ( Рис. S3 ). В связи с этим мы пришли к выводу, что при использовании мультитканевого набора данных прогнозные показатели улучшатся, если для расчета вероятности возраста будет учитываться больше CpG в расчете на одну клетку. Таким образом, мы использовали мультитканевый предиктор scAge с 2000 CpG, профилированным на одну клетку. Эта модель показала уменьшение точности по сравнению с моделью печени, коэффициент корреляции Пирсона 0,63 (Spearman rho = 0,72) и среднюю и медианную абсолютные ошибки 6,29 и 4,4 месяца, соответственно ( рис. 2f ). Интересно, что модель с несколькими тканями предсказывала, что возраст одной клетки в каждой группе близок к максимальному возрасту, который мы обозначили при запуске алгоритма. Мы интерпретируем эти наблюдения как траекторию ускоренного старения (т.е. ускоренного старения) некоторых клеток из популяции, что еще больше подчеркивает гетерогенность эпигенетического старения в отдельных клетках. Удаление обоих выбросов в моделях печени и нескольких тканей привело к значительному повышению точности прогноза со значениями r Пирсона 0,95 и 0,9 соответственно ( рис. S4 ).
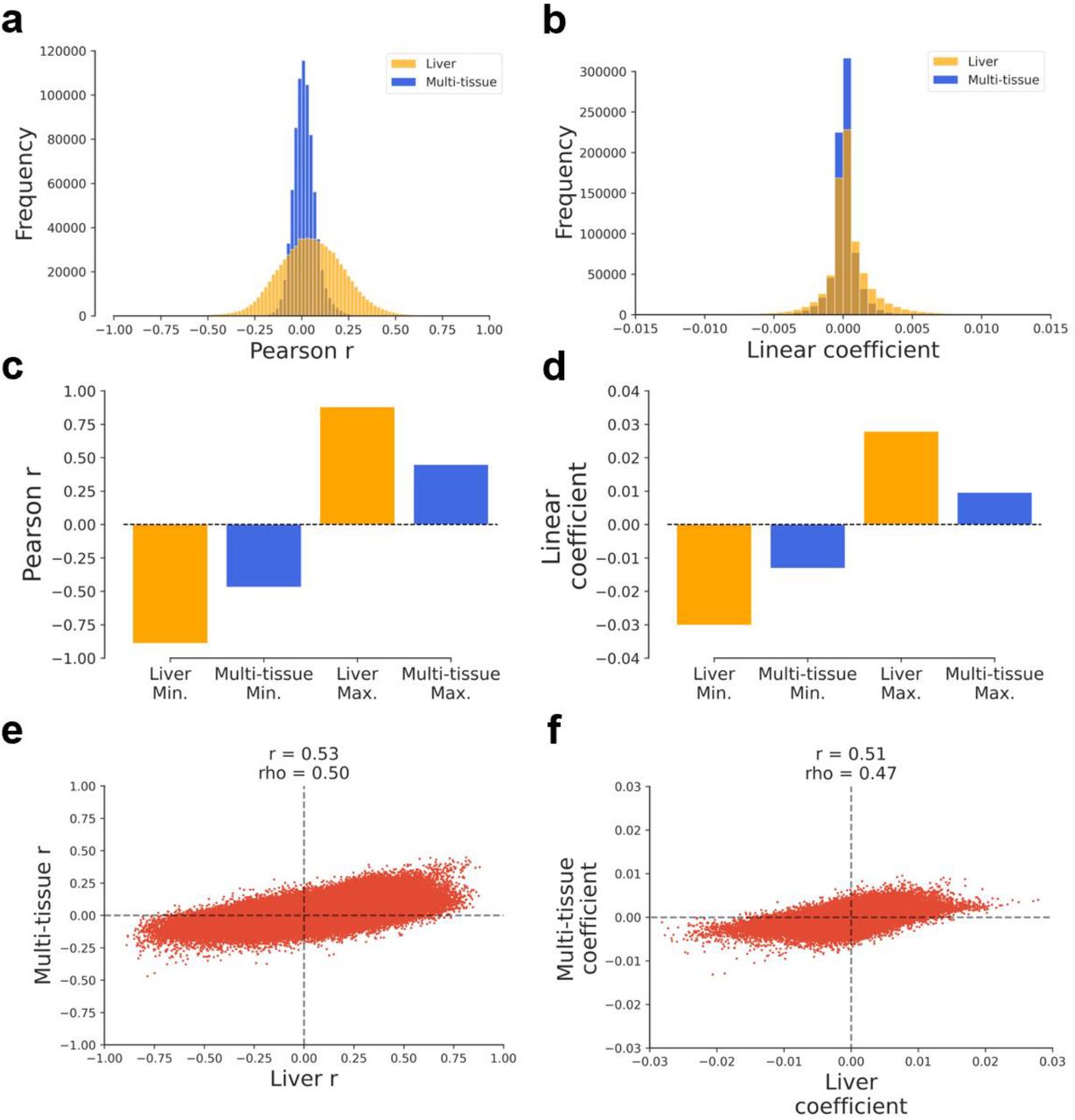 Рисунок S3:Показатели корреляции и регрессии различаются между наборами обучающих данных Распределение коэффициента Пирсона r (a) и коэффициентов линейной регрессии (b) для наборов обучающих данных для печени и мультитканевого. Минимальный и максимальный коэффициенты Пирсона r (c) и линейной регрессии (d) для наборов данных тренировки печени и нескольких тканей. Связь между наборами обучающих данных мультитканевым и печени Pearson r (e) и коэффициентами линейной регрессии (f) .
Рисунок S3:Показатели корреляции и регрессии различаются между наборами обучающих данных Распределение коэффициента Пирсона r (a) и коэффициентов линейной регрессии (b) для наборов обучающих данных для печени и мультитканевого. Минимальный и максимальный коэффициенты Пирсона r (c) и линейной регрессии (d) для наборов данных тренировки печени и нескольких тканей. Связь между наборами обучающих данных мультитканевым и печени Pearson r (e) и коэффициентами линейной регрессии (f) .
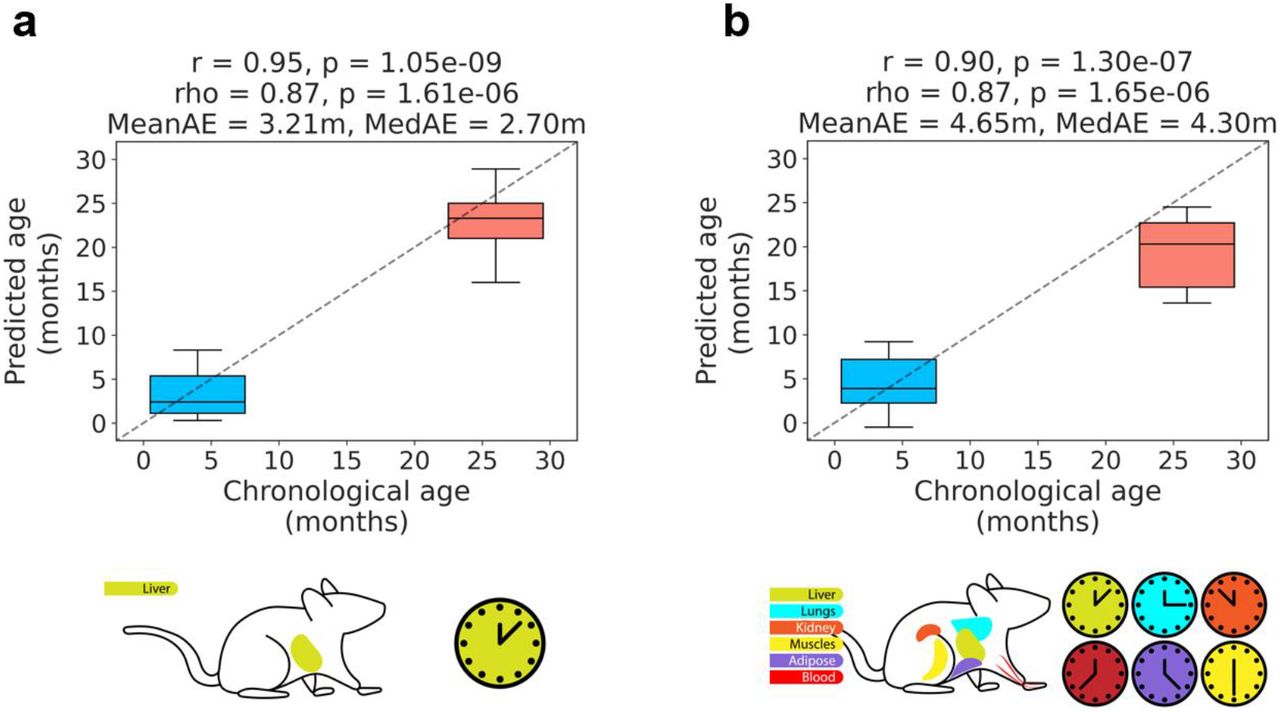 Рисунок S4:Точность прогнозирования масштабов повышается с удалением выбросов Прогнозы эпигенетического возраста для гепатоцитов печени с удалением одного выброса (ускоренное старение) на группу (SRR3136664 для молодых и SRR3136629 для старых) для моделей scAge печени (a), и мультитканевой (b) .
Рисунок S4:Точность прогнозирования масштабов повышается с удалением выбросов Прогнозы эпигенетического возраста для гепатоцитов печени с удалением одного выброса (ускоренное старение) на группу (SRR3136664 для молодых и SRR3136629 для старых) для моделей scAge печени (a), и мультитканевой (b) .
Интересно, что прогностические метрики этих двух моделей по данным печени варьировались в зависимости от количества CpG, включенных в расчет общего правдоподобия, в результате чего включение слишком малого или слишком большого количества CpG приводило к снижению точности предсказания ( рис. S5 ). Когда использовалось слишком мало CpG, было недостаточно индивидуальных вероятностей для расчета точного прогноза возраста ( рис. S5a-b ). Однако включение слишком большого количества CpG также привело к небольшому снижению точности прогнозов моделей ( рис. S5c-d). Поскольку наш алгоритм ранжировал CpG на основе того, как они соотносятся с возрастом, мы предлагаем, чтобы включение большего количества CpG с более низким рейтингом вносило шум в прогноз, тем самым снижая общую точность. Следует отметить, что точность прогнозов не показала значимой связи с количеством общих CpG между любой отдельной клеткой и обучающим набором данных ( рис. S6 ). Это означает, что наш метод устойчив к относительно низкому охвату отдельных клеток.
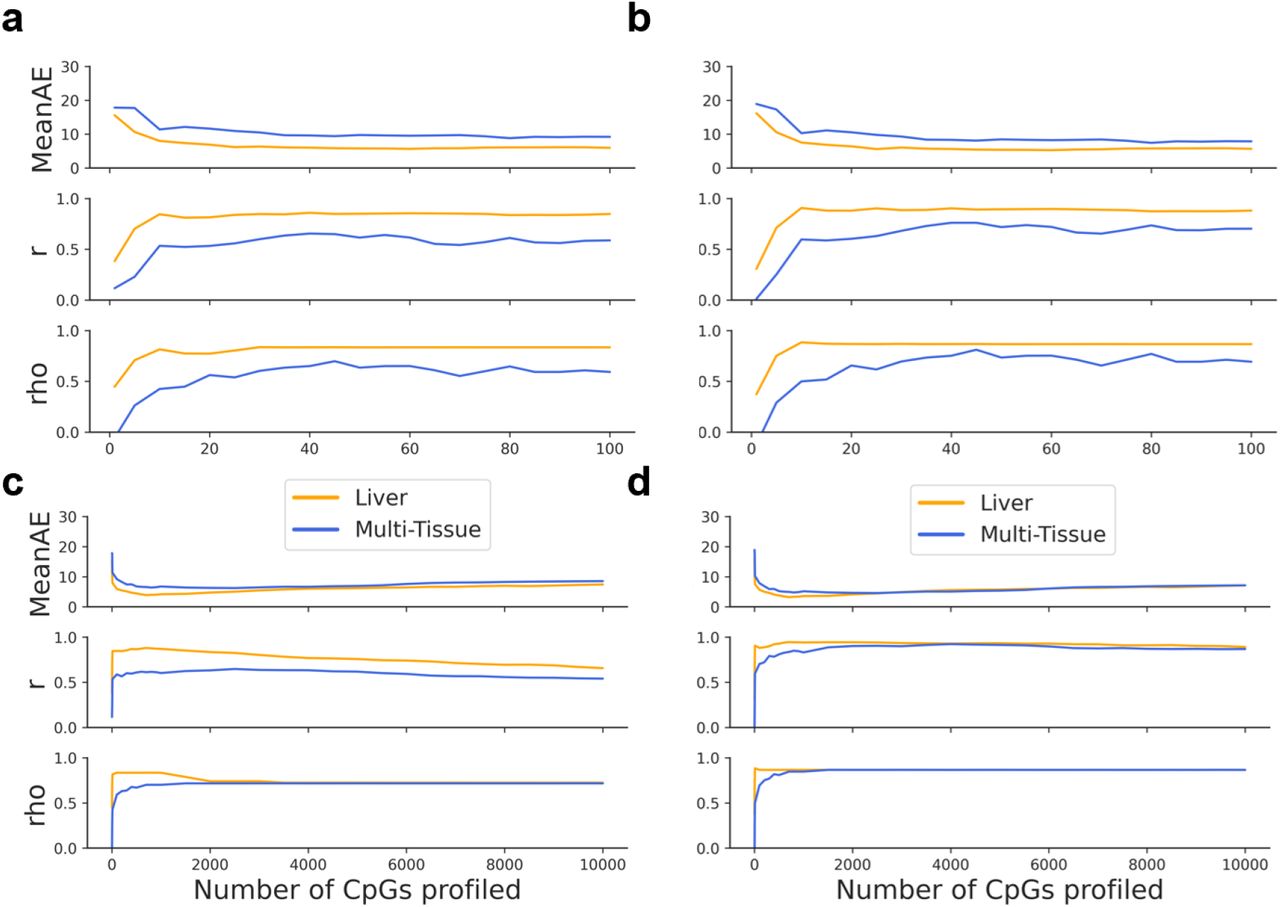 Рисунок S5: Количество CpG, профилированных в scAge, влияет на прогнозные показатели Прогностические показатели SCAge (MeanAE: средняя абсолютная ошибка, r: коэффициент корреляции Пирсона, rho: коэффициент корреляции Спирмена) для гепатоцитов варьируются в зависимости от количества CpG, включенных в алгоритм. Увеличение CpG, профилированных с 1 до 100 с шагом 5, обычно улучшает точность прогноза с включенными выбросами (а) или с удаленными выбросами (б) . Увеличение профилей CpG до 10000 на клетку с шагом 500 показывает, что промежуточное количество CpG приводит к наивысшей точности прогноза, как с включенными выбросами (c), так и с удаленными выбросами (d) .
Рисунок S5: Количество CpG, профилированных в scAge, влияет на прогнозные показатели Прогностические показатели SCAge (MeanAE: средняя абсолютная ошибка, r: коэффициент корреляции Пирсона, rho: коэффициент корреляции Спирмена) для гепатоцитов варьируются в зависимости от количества CpG, включенных в алгоритм. Увеличение CpG, профилированных с 1 до 100 с шагом 5, обычно улучшает точность прогноза с включенными выбросами (а) или с удаленными выбросами (б) . Увеличение профилей CpG до 10000 на клетку с шагом 500 показывает, что промежуточное количество CpG приводит к наивысшей точности прогноза, как с включенными выбросами (c), так и с удаленными выбросами (d) .
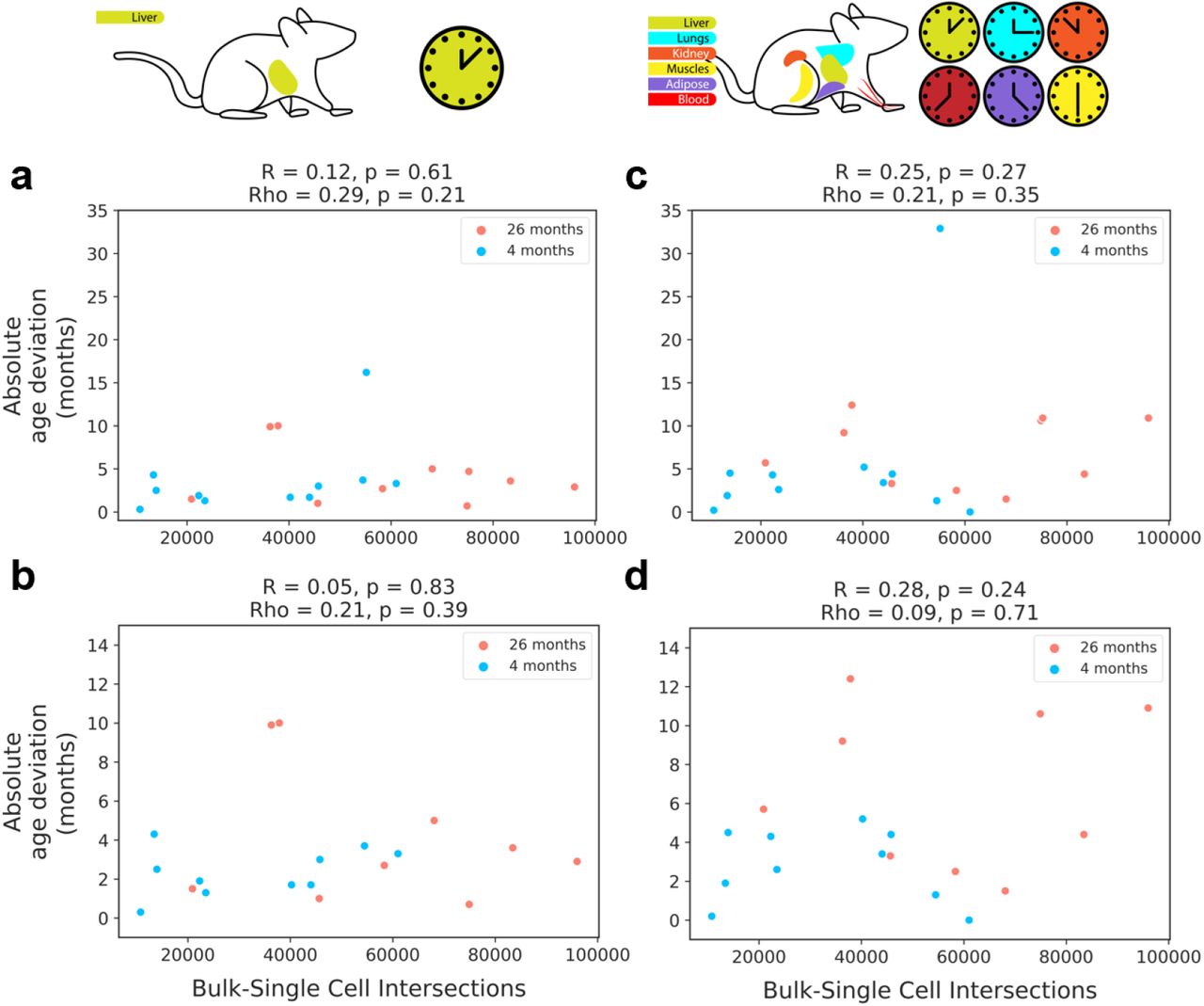 Рисунок S6: Покрытие CpG оказывает минимальное влияние на точность прогнозов Связь между абсолютным возрастным отклонением (абсолютное значение разницы между прогнозируемым возрастом в отдельной клетке и хронологическим возрастом ткани происхождения) и количеством обычных одноклеточных / массовых CpG с использованием часов scAge печени с выбросами (a) или с двумя удаленными выбросами (b) и с использованием часов шкалы scAge для нескольких тканей с выбросами (c) или с удаленными выбросами (d).
Рисунок S6: Покрытие CpG оказывает минимальное влияние на точность прогнозов Связь между абсолютным возрастным отклонением (абсолютное значение разницы между прогнозируемым возрастом в отдельной клетке и хронологическим возрастом ткани происхождения) и количеством обычных одноклеточных / массовых CpG с использованием часов scAge печени с выбросами (a) или с двумя удаленными выбросами (b) и с использованием часов шкалы scAge для нескольких тканей с выбросами (c) или с удаленными выбросами (d).
Часы SCAge предсказывают, что возраст эмбриональных фибробластов близок к нулю
Мы также применили scAge к 5 эмбриональным фибробластам мыши (MEF), включенным в тот же набор данных. MEF имели значительно более высокий охват, чем гепатоциты, благодаря улучшенному качеству ДНК, которое стало результатом более мягкого процесса выделения по сравнению с клетками печени ( рис. S2a ) ( Gravina et al., 2016 ). Кроме того, среднее метилирование в MEF было ниже, чем у гепатоцитов ( рис. 2b ). scAge, обученный либо на печени, либо на наборах данных с несколькими тканями, предсказал, что эпигенетический возраст MEF будет около 0 ( рис. 2g-h). Несмотря на то, что эти клетки находятся в культуре, оказалось, что они сохраняют эпигенетическую информацию о возрасте эмбриона. В целом, наши результаты показывают, что scAge, обученный на наборе данных печени или нескольких тканей, может точно воспроизводить хронологический возраст ткани происхождения в отдельных гепатоцитах и эмбриональных фибробластах.
Мышечные стволовые клетки демонстрируют минимальное эпигенетическое старение
Для дальнейшего изучения применимости scAge мы применили его к данным о молодых и старых мышечных стволовых клетках ( Hernando-Herraez et al., 2019 ). Этот набор данных состоял из 275 отдельных клеток от 6 доноров, в том числе 4 молодых (1,5 месяца) и 2 старых животных (26 месяцев). Из-за технической вариабельности методологии scBS только 185 клеток (67%) имели более 1 миллиона покрытых CpG ( рис. S2b ). Среднее метилирование между молодыми и старыми клетками было сопоставимым.
Когда мы применили к этим мышечным стволовым клеткам тот же мультитканевый scAge, который профилирует 2000 CpG, эпигенетический возраст молодых клеток составил в среднем 9,5 недель, что примерно соответствует их хронологическому возрасту. Интересно, что старые мышечные стволовые клетки показали значительное эпигенетическое увеличение возраста, но всего на несколько недель, со средним прогнозируемым возрастом 18,3 недели ( рис. 3a-b ). Эти результаты согласуются с предыдущим анализом, в котором изучался эпигенетический возраст этих клеток с помощью подхода псевдо-массового группирования с использованием мышечных часов, обученных с помощью обычных методов эластичной сети ( Hernando-Herraez et al., 2019 ; Reizel et al., 2015 ; Stubbs и др., 2017). В целом, наши результаты согласуются с ранее сообщенной динамикой эпигенетического старения мышечных стволовых клеток мышей, но дают разрешение данных на уровне отдельной клетки.
Единичные эмбриональные стволовые клетки демонстрируют низкий эпигенетический возраст
Затем мы попытались оценить scAge на наиболее распространенном типе общедоступных наборов данных о метилировании отдельных клеток: профилирующих эмбриональную ткань. Эмбриональные стволовые клетки (ESC) и их аналоги на основе индуцированных плюрипотентных стволовых клеток (IPSC) обычно показывают очень низкий прогнозируемый эпигенетический возраст, стремящийся к 0 ( Horvath, 2013 ; Meer et al., 2018 ; Petkovich et al., 2017 ). Чтобы проверить нашу модель, мы изучили 3 набора данных об эмбриональных стволовых клетках и родственных тканях ( Angermueller et al., 2016 ; Clark et al., 2018 ; Smallwood et al., 2014 ). Клетки из этих исследований показали переменный охват, и мы выборочно отфильтровали
те клетки, в которых было охвачено не менее 1 миллиона CpG, чтобы улучшить согласованность между наборами данных (Рис. S2c ). Важно отметить, что ESC культивировали либо в традиционных условиях сыворотки, либо в бессывороточной среде с добавлением коктейля «2i» ингибиторов MEK и GSK3β. Ранее было показано, что культивирование клеток в среде 2i управляет глобальным гипометилированием в ESC, создавая эпигенетические профили, соответствующие мигрирующим примордиальным зародышевым клеткам ( Ficz et al., 2013 ).
Как и ожидалось, мы наблюдали значительное гипометилирование среди клеток 2i в исследованиях Angermueller et al. и Smallwood et al. ( рис. 3в ). Ооциты MII у Smallwood et al. показали среднее глобальное метилирование сопоставимое с клетками 2i , а эмбриоидные тела, полученных из ESC у Clark et al. показали в среднем наибольшее среднее метилирование, значительно выше, чем у ESC, выращенных в сыворотке и 2i ( рис. 3c ). Мы применили нашу печеночную и мультитканевую модели scAge ко всем отфильтрованным клеткам из этих трех исследований и наблюдали стабильно низкий прогнозируемый возраст для всех проанализированных типов клеток ( рис. 3d-e ). При использовании модели для печени, ESC, культивируемые в сыворотке, показали эпигенетический возраст около 0, в то время как ESC 2i показали значительно более высокий прогнозируемый эпигенетический возраст (Рис. 3г ). Однако мультитканевый предиктор возраста scAge показал противоположную тенденцию ( рис. 3e ). Кроме того, мультитканевая модель продемонстрировала большую дисперсию и более экстремальные прогнозы по сравнению с моделью печени во всех трех наборах данных. Мы предполагаем, что на мультитканевых наборах данных отображается менее устойчивая линейная зависимость от возраста, в результате чего повышенные различия в уровнях метилирования между различными тканями приводят к менее согласованным прогнозам ( рис. S3).). Эмбриоидные тельца, полученные из выращенных в сыворотке ESC, продемонстрировали более высокий возраст при использовании обоих часов, намекая на то, что инициация немодулированных сигналов дифференцировки в трех зародышевых листках быстро вызывает заметное увеличение эпигенетического возраста клеток. В целом, наши результаты предсказали, что эпигенетический возраст эмбриональных стволовых клеток будет близок к нулю, при этом обнаружились существенные различия в зависимости от конкретных условий культивирования.
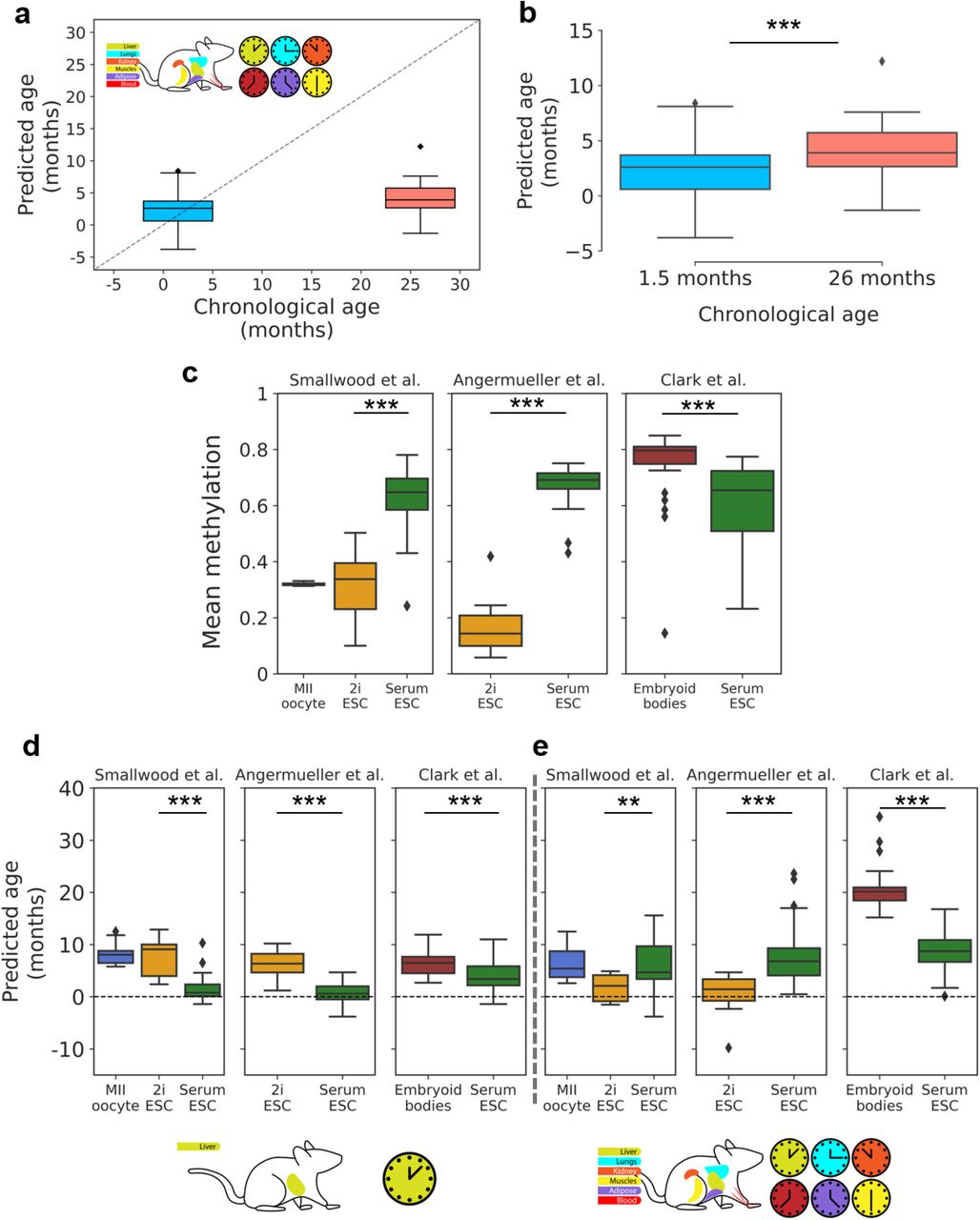 Рисунок 3:scAge выявляет четкие эпигенетические траектории старения стволовых клеток Эпигенетические прогнозы возраста мышечных стволовых клеток с использованием мультитканевых часов scAge, построенные в зависимости от хронологического возраста (а) или помещенные рядом (б) . *** обозначает p & 0,001. c) Среднее метилирование ооцитов, эмбриональных стволовых клеток (ESC) в условиях культивирования 2i или сыворотки и эмбриоидных телец. Клетки сгруппированы по исследованиям. *** означает p <0,001. Прогнозирование эпигенетического возраста для всех типов клеток в трех эмбриональных исследованиях с помощью scAge часов печени (d) или мультитканевых scAge часов (e) . ** обозначает p & 0,01, *** обозначает p <0,001.
Рисунок 3:scAge выявляет четкие эпигенетические траектории старения стволовых клеток Эпигенетические прогнозы возраста мышечных стволовых клеток с использованием мультитканевых часов scAge, построенные в зависимости от хронологического возраста (а) или помещенные рядом (б) . *** обозначает p & 0,001. c) Среднее метилирование ооцитов, эмбриональных стволовых клеток (ESC) в условиях культивирования 2i или сыворотки и эмбриоидных телец. Клетки сгруппированы по исследованиям. *** означает p <0,001. Прогнозирование эпигенетического возраста для всех типов клеток в трех эмбриональных исследованиях с помощью scAge часов печени (d) или мультитканевых scAge часов (e) . ** обозначает p & 0,01, *** обозначает p <0,001.
Анализ отдельных клеток предполагает событие омоложения во время гаструляции у мыши.
Затем мы исследовали набор данных, профилирующий гаструляцию у мышей при отдельноклеточном разрешении ( Argelaguet et al., 2019 ). Эти данные состояли из 758 отдельных клеток, выделенных из эмбрионов мышей по времени (E) от 4,5 до 7,5 эмбриональных дней. Мы отфильтровали данные, чтобы сохранить клетки с охватом не менее 500 000 CpG, в результате чего получили окончательный набор данных из 495 клеток ( рис. S2d ). Интересно, что среднее глобальное метилирование сильно варьировало во время этого раннего периода гаструляции мышей, при этом клетки E4.5 обнаруживают значительное гипометилирование по сравнению с остальными тремя стадиями развития ( Рис. 4a ). Эта тенденция глобального метилирования указывает на связь между ESC, выращенными в условиях 2i, и клетками эмбрионов E4.5.
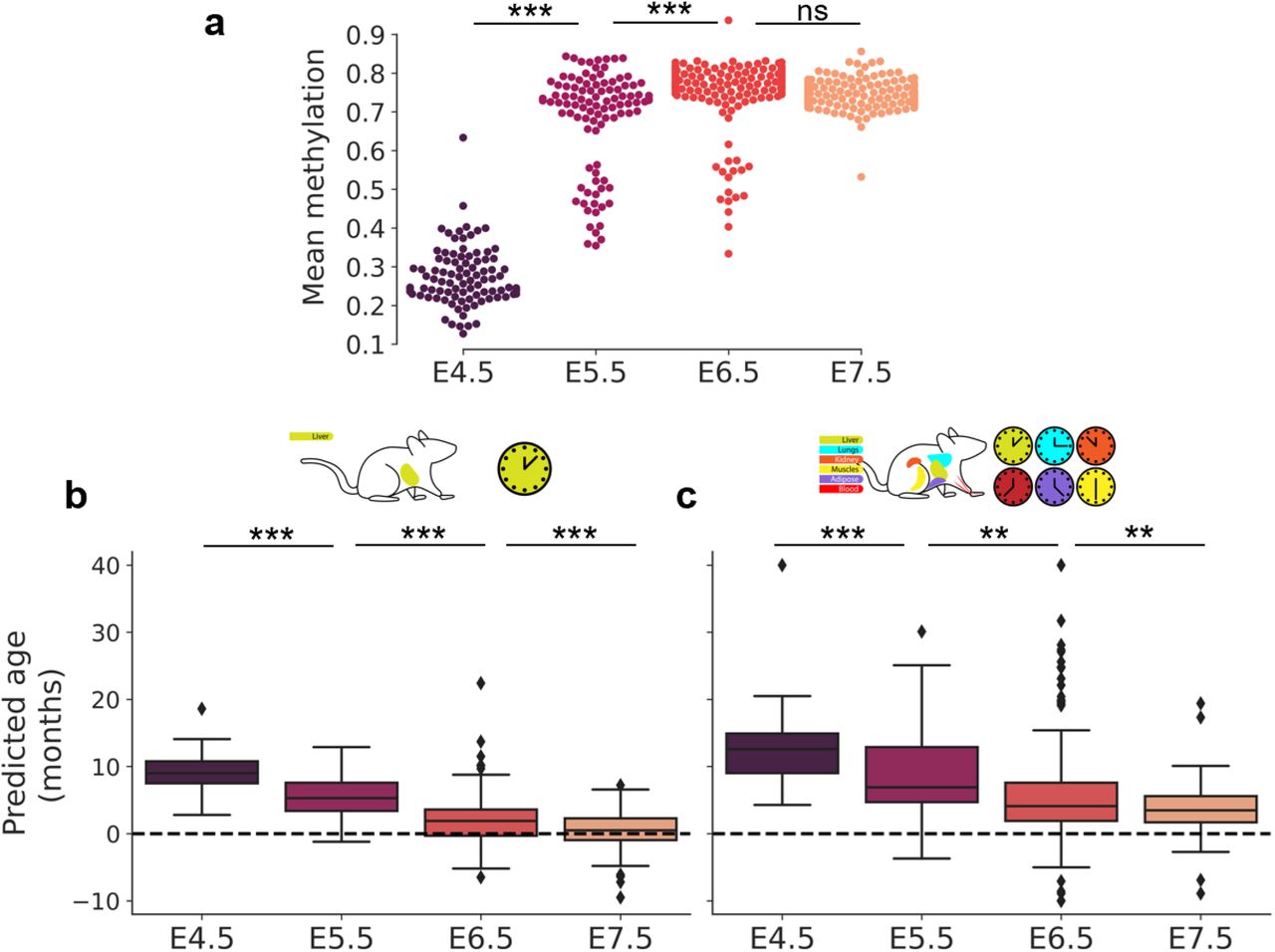 Рисунок 4: SCAge предполагает событие омоложения во время гаструляции а) Среднее метилирование среди клеток E4.5 - E7.5. *** означает p <0,001. Прогнозирование эпигенетического возраста для отдельных эмбриональных клеток на протяжении гаструляции с помощью scAge часов печени (b) и мультитканевых scAge часов (c) . ** обозначает p <0,01, *** обозначает p <0,001.
Рисунок 4: SCAge предполагает событие омоложения во время гаструляции а) Среднее метилирование среди клеток E4.5 - E7.5. *** означает p <0,001. Прогнозирование эпигенетического возраста для отдельных эмбриональных клеток на протяжении гаструляции с помощью scAge часов печени (b) и мультитканевых scAge часов (c) . ** обозначает p <0,01, *** обозначает p <0,001.
Недавно было высказано предположение, что эмбриогенез может характеризоваться начальным снижением биологического возраста до точки, называемой «нулевой точкой», после чего формально начинается старение организма ( Гладышев, 2021 ). В соответствии с этой идеей недавнее применение эпигенетических часов к массивным образцам выявило значительное снижение биологического возраста (т.е. омоложение) на ранних стадиях эмбриогенеза с последующим увеличением на более поздних стадиях ( Kerepesi et al., 2021 ). Это открытие также согласуется с представлением о том, что накопление повреждений неизбежно происходит в течение жизни организма, даже в половых клетках. Таким образом, считается, что событие омоложения происходит в середине эмбриогенеза, чтобы обеспечить непрерывное воспроизводство новых биологически молодых особей.
Чтобы исследовать эту идею на уровне отдельных клеток, мы применили scAge часы печени и мультитканевые к отдельным эмбриональным клеткам на четырех исследуемых стадиях развития. scAge часы печени показали устойчивое и значительное снижение среднего прогнозируемого возраста в период от E4,5 до E7,5, причем последнее значение возраста было около 0 ( рис. 4b ). Мультитканевые scAge часы показали идентичную тенденцию, хотя и с несколько повышенным и более изменчивым прогнозируемым возрастом ( рис. 4c).). Вместе эти результаты показывают, что событие омоложения происходит в середине эмбриогенеза и что отдельные клетки могут быть омоложены естественными способами. Примечательно, что самый низкий эпигенетический возраст единичных клеток приблизительно соответствует стадии гаструляции и связан с гиперметилированием, предполагая, что для омоложения клеток важно как деметилировать, так и впоследствии реметилировать геном.
ОБСУЖДЕНИЕ
В этой работе мы представляем scAge, вероятностную модель для определения эпигенетического возраста отдельных клеток. Наш метод использует данные о массовом метилировании для обучения моделей линейной регрессии, которые предсказывают уровни метилирования, учитывая исключительно возраст в качестве входных данных. На основе этих одномерных моделей мы вычисляем апостериорную вероятность наблюдения неметилированного или метилированного состояния в отдельной клетке. Используя выбранную фракцию возрастных CpG и связанных с ними вероятностей, мы вычисляем вероятность того, что клетка происходит из ткани определенного хронологического возраста, и регистрируем возраст максимальной вероятности в качестве конечного предиктора эпигенетического возраста. Этот подход решает проблемы разреженности и неравномерного покрытия профилей метилирования отдельных клеток, что препятствует попыткам оценки эпигенетического возраста в отдельных клетках. Действительно, все предыдущие эпигенетические часы требуют определенных наборов сайтов CpG для их применения, что невозможно в случае одиночных клеток.
Этот метод позволяет точно прогнозировать возраст отдельных гепатоцитов и эмбриональных фибробластов мыши с высоким разрешением на моделях, обученных либо на печени, либо на мультитканевых наборах данных. Кроме того, мы демонстрируем согласованность между нашей моделью и предыдущей работой со стволовыми клетками мышей, которые демонстрируют ослабленное эпигенетическое старение по сравнению с их хронологическим возрастом. Мы также обнаружили, что, хотя, ESC обычно имеют низкий эпигенетический возраст, возраст различается в зависимости от условий культивирования. Наконец, наши данные предоставляют дополнительное доказательство гипотезы старения «нулевой точки», показывая очень значимое и устойчивое снижение эпигенетического возраста отдельных клеток во время гаструляции.
Несмотря на прогресс в эпигенетическом профилировании возраста отдельных клеток, существуют различные возможности для улучшения. Во-первых, здесь предполагалось, что состояния бинарного метилирования CpG независимы друг от друга, поскольку в предыдущих работах говорилось, что при анализе единичных чтений из совокупных образцов это имело место ( Han, Franzen, et al., 2020 ; Han, Nikolić, et al. др., 2020). Однако более тщательный анализ этого поведения, особенно в отдельных клетках, может выявить биологические идеи, предполагающие более сложные отношения. Кроме того, использование только линейной регрессии может быть неоптимальным при рассмотрении потенциально разнообразного набора математических ассоциаций, которые лучше всего моделируют уровни метилирования CpG и возраст. Наконец, остается исследовать, как индивидуальные траектории старения клеток меняются со временем и как они передаются во время таких событий, как деление клеток.
Взятые вместе, эти результаты предполагают драматические последствия в отношении эпигенетического старения. Мы обнаружили, что совокупность нескольких прогнозов по отдельным клеткам дает точный средний показатель возраста конкретной ткани. Однако этот одноклеточный подход одновременно обнаруживает глубокую неоднородность траекторий старения отдельных клеток. Это говорит о том, что все клетки в ткани стареют, но часы, вероятно, тикают независимо в пределах одной клетки. В свою очередь, мы предполагаем, что некоторые клетки подвергаются ускоренному или замедленному эпигенетическому старению, что ранее было невозможно установить ( рис. 5а).). Кроме того, этот метод может найти широкое клиническое применение для человеческих соматических, зародышевых и раковых клеток, поскольку с помощью этого подхода можно различать и картировать «молодые» и «старые» клетки в гетерогенной ткани ( рис. 5b ). В целом, мы представляем здесь первый метод определения эпигенетического возраста в отдельных клетках с далеко идущим потенциалом во всем что касается старения.
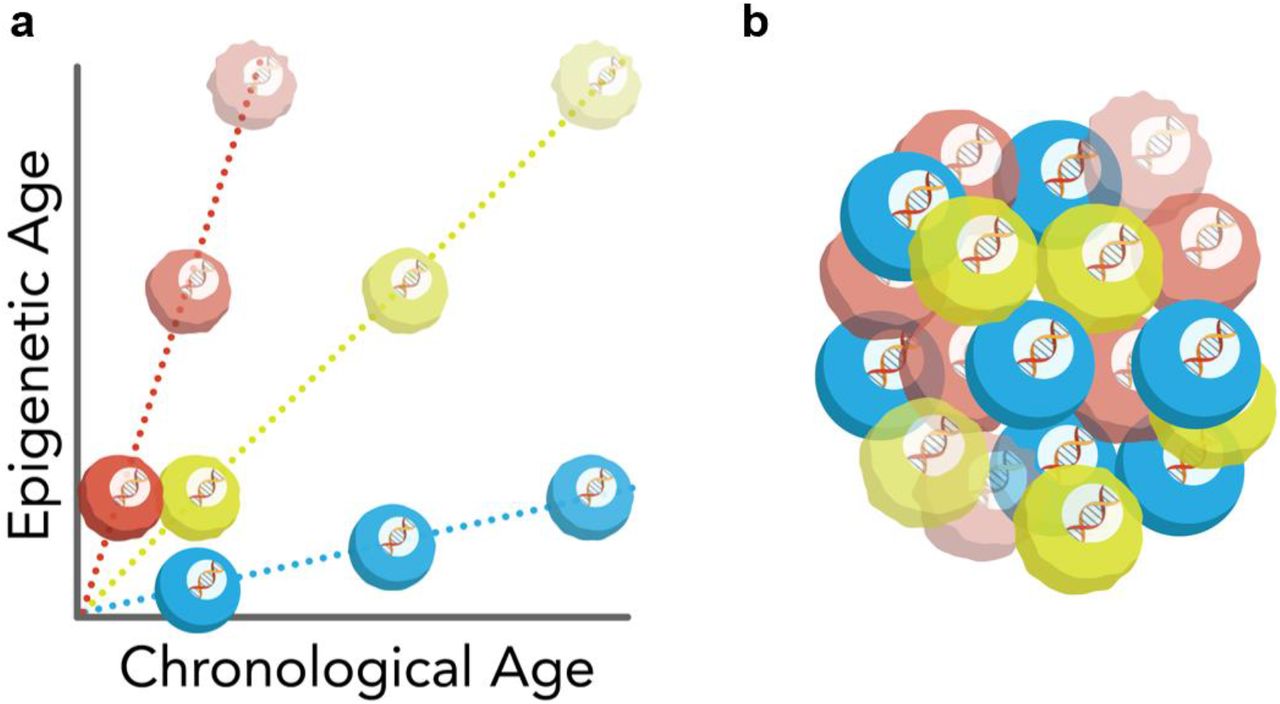 Рисунок 5: Модели старения в отдельных клетках а) Схематическая модель траекторий старения одиночных клеток. Некоторые клетки (красные) показывают ускоренное эпигенетическое старение, в то время как другие клетки (синие) показывают замедленное эпигенетическое старение по сравнению с эталонной популяцией клеток (зеленый). б) Схематическое изображение неоднородности эпигенетического старения в популяции клеток, которая состоит из клеток с ускоренной (красный), замедленной (синий) и средней (зеленый) траекториями старения.
Рисунок 5: Модели старения в отдельных клетках а) Схематическая модель траекторий старения одиночных клеток. Некоторые клетки (красные) показывают ускоренное эпигенетическое старение, в то время как другие клетки (синие) показывают замедленное эпигенетическое старение по сравнению с эталонной популяцией клеток (зеленый). б) Схематическое изображение неоднородности эпигенетического старения в популяции клеток, которая состоит из клеток с ускоренной (красный), замедленной (синий) и средней (зеленый) траекториями старения.
МЕТОДЫ
Обработка данных отдельных клеток
Для Gravina et al. данные секвенирования были загружены из SRA под регистрационным номером SRA344045 ( Gravina et al., 2016 ). В этом случае данные секвенирования были предварительно обрезаны перед нанесением на SRA. Обрезанные последовательности были сопоставлены с геномом mm10 / GRCm38.p6 с использованием Bismark v0.22.3 с опцией - non_directional , как это было предложено в Руководстве пользователя Bismark v0.21.0 для препаратов библиотеки Zymo Pico-Methyl scWGBS. Чтения были дедуплицированы, а уровни метилирования для сайтов CpG были извлечены с помощью Bismark ( Krueger & Andrews, 2011 ).
Для Hernando-Herraez et al., Angermueller et al., Clark et al., Smallwood et al. И Argelaguet et al. исследований, обработанные файлы покрытия, содержащие извлеченные уровни метилирования, сгенерированные Bismark, были загружены непосредственно из базы данных GEO под номерами доступа GSE121436, GSE68642, GSE109262, GSE56879 и GSE121690, соответственно ( Angermueller et al., 2016 ; Argelaguet et al., 2019 ; Clark et al., 2018 ; Hernando-Herraez et al., 2019 ; Smallwood et al., 2014 ).
Затем все файлы покрытия были дополнительно обработаны для масштабирования уровня метилирования до соотношения между [0, 1]. Хотя профили метилирования отдельных клеток были почти полностью бинарными, технические соображения, такие как систематическая ошибка амплификации ПЦР, привели к некоторым промежуточным значениям метилирования. Неопределенные вызовы метилирования 0,5 были удалены перед последующим анализом. Оставшиеся значения метилирования округляли до 0 или 1. Геномные позиции на 19 аутосомах мыши были сохранены для анализа, чтобы частично минимизировать влияние пола на исследование. Покрытие интерпретировалось как общее количество покрытых метилированных и неметилированных цитозинов на обеих цепях ДНК. Среднее метилирование в отдельных клетках вычисляли как среднее всех наблюдаемых состояний бинарного метилирования.
Массовая обработка данных
Чтобы создать массивные эталонные наборы данных для оценки линейной зависимости между возрастом и уровнем метилирования, мы загрузили обработанные данные RRBS исследования Thompson et al. , депонированные в базе данных GEO под инвентарным номером GSE120132 ( Thompson et al., 2018). Этот набор данных состоял из 549 полных образцов из печени, легких, крови, почек, жировой и мышечной тканей в возрасте от 1 месяца до 21 месяца. Фракции метилирования были приняты как число прочтений, подтверждающих метилированный статус для CpG, по сравнению с общим числом чтений, которые покрывают этот CpG. Чтобы максимизировать точность уровней массового метилирования при одновременном сохранении как можно большего количества сайтов, были сохранены только сайты CpG, для которых 90% образцов имели как минимум 5-кратное покрытие. Это привело к окончательной мульти-тканевой матрице из 549 образцов 748 955 положительных цепей CpG (только аутосомные хромосомы) с некоторыми пропущенными значениями. Отсюда была создана отдельная матрица только для печени, содержащая 60 образцов печени с возрастом от 2 до 20 месяцев на основе того же набора из 748 955 CpG. Корреляции Пирсона с возрастом рассчитывались с использованиемcorrwith из пакета pandas . Линейные регрессии были рассчитаны с использованием функции LinearRegression как части пакета sklearn .
Вероятностные одноклеточные часы
На разработку алгоритма для определения эпигенетического возраста отдельных клеток нас вдохновила недавняя работа лаборатории Вагнера по анализу возраста при секвенировании отдельных бисульфитных ампликонов со штрих-кодом (BBA-seq), считанных из массивных образцов ( Han, Franzen, et al. , 2020 ; Хан, Николич и др., 2020 ). Сначала мы вычислили линейные регрессии для каждой CpG, включенной в обучающий набор данных, в форме: 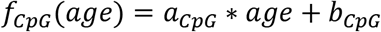 где возраст в месяцах - независимая переменная, f CpG ( возраст ) - прогнозируемый средний уровень метилирования, а a и b - коэффициент линейной регрессии и точка пересечения, соответственно ( Fig. 1c ). Мы также рассчитали коэффициент корреляции Пирсона с возрастом для каждого CpG в обучающей выборке.
где возраст в месяцах - независимая переменная, f CpG ( возраст ) - прогнозируемый средний уровень метилирования, а a и b - коэффициент линейной регрессии и точка пересечения, соответственно ( Fig. 1c ). Мы также рассчитали коэффициент корреляции Пирсона с возрастом для каждого CpG в обучающей выборке.
Затем мы сопоставили CpG, включенные в набор обучающих данных, с CpG в любой данной отдельной клетке, получив серию из n CpG, которые присутствуют как в групповых, так и в индивидуальных профилях отдельных клеток ( рис. 1d ). Мы отфильтровали эти n CpG на основе абсолютного значения их корреляции с возрастом, выбрав (в модели печени) 700 CpG с наибольшей абсолютной корреляцией Пирсона и (в модели с множеством тканей) 2000 CpG с наибольшей абсолютной корреляцией Пирсона. . Это количество CpG для включения в каждую модель было определено in silico.на основе тех, которые генерировали наиболее оптимальные метрики точности с использованием Gravina et al. набор данных в качестве ориентира. Следует отметить, что можно использовать различное количество CpG с минимальными колебаниями в эпигенетических прогнозах возраста ( рис. S5 ).
Для каждого выбранного CpG в клетке мы перебирали возраст с шагом 0,1 месяца от минимального возраста до максимального параметра возраста. Используя формулу линейной регрессии, рассчитанную для отдельного CpG, мы вычислили f CpG ( возраст ), который обычно находится в диапазоне от 0 до 1. Если это значение лежит за пределами диапазона (0, 1), оно вместо этого заменяется на 0,001 или 0,999 в зависимости от близости к любому значению. Далее мы предполагаем, что вероятность наблюдения метилированной одиночной клетки, происходящей из ткани данного возраста, приблизительно равна f CpG ( age ), то есть Pr CpG ( age ) = f CpG ( age).). Тогда вероятность того, что одна клетка метилирована в этом CpG, равна Pr CpG ( age ), и наоборот, вероятность того, что одна клетка не метилирована в этом CpG, равна 1 - Pr CpG ( age ). Это обеспечивает зависящую от возраста вероятность P для каждого общего CpG, сохраненного в алгоритме ( рис. 1e ).
Произведение каждой из этих вероятностей будет общей вероятностью наблюдаемого паттерна метилирования: 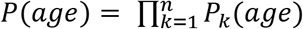 где k представляет собой отдельные CpG. Наша цель - найти максимум этого продукта среди разных возрастов (т.е. найти наиболее вероятный возраст для наблюдения этого конкретного паттерна метилирования). Для этого мы взяли сумму по CpG логарифма вероятностей (чтобы избежать ошибок потери значимости во время вычислений). Это дает нам
где k представляет собой отдельные CpG. Наша цель - найти максимум этого продукта среди разных возрастов (т.е. найти наиболее вероятный возраст для наблюдения этого конкретного паттерна метилирования). Для этого мы взяли сумму по CpG логарифма вероятностей (чтобы избежать ошибок потери значимости во время вычислений). Это дает нам 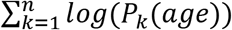 для каждой возрастной ступени. Эти логарифмические суммы обеспечивают показатель вероятности для каждого возраста, когда одна клетка происходит из основной ткани этого возраста. Наконец, мы выбираем возраст максимального правдоподобия в качестве предиктора возраста для одной клетки.
для каждой возрастной ступени. Эти логарифмические суммы обеспечивают показатель вероятности для каждого возраста, когда одна клетка происходит из основной ткани этого возраста. Наконец, мы выбираем возраст максимального правдоподобия в качестве предиктора возраста для одной клетки.
Вычислительный и статистический анализ
Все анализы проводились с использованием Python 3.8.3 со стандартным набором научных, математических и графических пакетов. Для обработки данных секвенирования использовались собственные сценарии bash. Для выполнения всех статистических тестов использовался t-критерий Велча, предполагающий неравные дисперсии. P-значения менее 0,05 были приняты как значимые. * Обозначает p <0,05, ** обозначает p <0,01, а *** обозначает p <0,001.
ВКЛАД АВТОРА
AT выполнил все анализы. AT и CK разработали метод и реализовали алгоритм. CK внесла свой вклад в предварительную обработку данных. AT и VNG написали рукопись при участии CK. Компания VNG разработала исследование и руководила работой.
НАЛИЧИЕ КОДА
Исходный код scAge будет доступен после публикации.
КОНКУРИРУЮЩИЕ ИНТЕРЕСЫ
AT, CK и VNG названы изобретателями в предварительной заявке на патент на scAge.
БЛАГОДАРНОСТИ
Мы благодарим Тиамат Фокс и Адит Гангули за помощь со схематическими изображениями. Мы также благодарим Марко Мариотти, Анастасию Шиндьяпину, Сун Хи Йим, Санг-Гу Ли, Дидака Сантесмассеса и Патрика Гриффина за полезное обсуждение. Поддерживается грантами NIA для VNG.
Опубликовано: 15 марта 2021г.
Авторы: Александр Трапп,Чаба Керепеси,Вадим Николаевич Гладышев
Оригинальная статья: Profiling epigenetic age in single cells
Перевод Ник Сестрин