Генная терапия. Такое, кажется, уже замусоленное до дыр словосочетание, так часто обсуждаемое, любимое во многих научно-популярных статьях. Возможно, это не очень хорошо, потому что нередко поверхностно усваиваемая людьми информация ведёт к недостатку глубокого понимания того, что стоит за этим понятием. Чтобы внести чуть больше света в это тёмное царство, мы и поговорим о некоторых методах генной терапии: что нужно для того, чтобы заменить дефекты генов, и как это выглядит на практике.
Генная терапия действительно является многообещающей индивидуализированной областью терапии ранее неизлечимых заболеваний, а потому так часто на слуху. С тех пор, как изучение фундаментальных основ жизни стало неразрывно связано с пониманием процессов на молекулярном и клеточном уровнях, возникло и желание вносить поправки в меж- и внутриклеточные взаимодействия, чтобы предотвратить развитие различных патологий, в том числе и наследственных — опосредованных генетическими нарушениями в клетках. Чтобы исправить последствия мутации в каком-либо гене, необходимо «протащить» терапевтический ген в целевые клетки, несущие мутантный ген, ответственный за развитие патологического процесса. Таким образом запускается физиологически необходимый синтез функционально полноценного белка, восполняющего вызванную мутацией недостаточность. Подобная концепция возникла в учёных умах в 70-х годах прошлого века, как только стало возможным клонирование генов, и было на примере клеточных линий млекопитающих in vitro показано, что искусственно размноженные последовательности нуклеотидов способны скорректировать патологический фенотип. И на сегодняшний день развитие стратегий генной терапии позволяет задумываться не только о лечении моногенных заболеваний, но и куда более сложных болезней, в основе которых лежат множественные мутации.
Достижение возобновления синтеза необходимого продукта какого-либо гена путём внесения терапевтического гена особенно значимо при рецессивных заболеваниях, когда мутация ведет к снижению или прекращению экспресии белка. В случае же аутосомно-доминантных мутаций, когда синтезируемый белок по структуре отличается от нормального и может оказывать токсическое действие, вероятно, простое внесения гена без дефекта проблемы не решит, так как нужно будет дополнительно предотвратить дальнейший синтез мутантного белка, например, при помощи малых интерферирующих РНК (siRNA).
Перед тем, как обратиться непосредственно к описанию введения гена в целевые клетки, важно отметить, что предварительно следует внести ген, к примеру, в бактериальные клетки, которые, задействуя собственные механизмы репликации, увеличивают количество копий терапевтического гена, т.е. многократно клонируют его. Генная последовательность, подготавливаемая для внесения в клетку, состоит из кодирующего участка ДНК (копия, клоновая ДНК), сопровождаемого регуляторными последовательностями, регулирующими транскрипцию и трансляцию кодирующей последовательности в принимающей клетке. Для того, чтобы данная конструкция оказалась в клетке, необходимы векторы, которые специфически подбираются в соответствии с тем, какой цели они служат (векторы могут быть клонирующими, челночными, про-/эукариотическими экспрессирующими, вирусными и т.д.) и в какие клетки какой ткани необходимо поместить заданный ген. Первое сообщение о создании транспортной системы для чужеродной ДНК, можно сказать, положившее начало генной инженерии, было сделано Стенли Коэном и двумя его коллегами в 1973 году: они «разрезали» две плазмиды с помощью фермента рестрикции EcoR-I, а затем «сшили» две плазмиды в одну посредством лигазы, получив тем самым гибридную молекулу ДНК. Каждая из исходных плазмид содержала по гену устойчивости к антибиотику, образованная рекомбинантная плазмида несла оба этих гена антибиотикорезистентности. Пятью годами позже данного успеха был создан первый плазмидный вектор, названный рВR322 (по фамилиям первооткрывателей — Боливара и Родригеза), отвечающий всем необходимым условиям для клонирования фрагмента ДНК. Этот вектор состоял из трёх частей: гена резистентности к тетрациклину (взят из плазмиды сальмонеллы pSC101), гена резистентности к ампициллину транспозона Tn3 и репликационного участка ori (место начала репликации — origin of replication) с прилежащими последовательностями плазмиды E. coli pMB1.
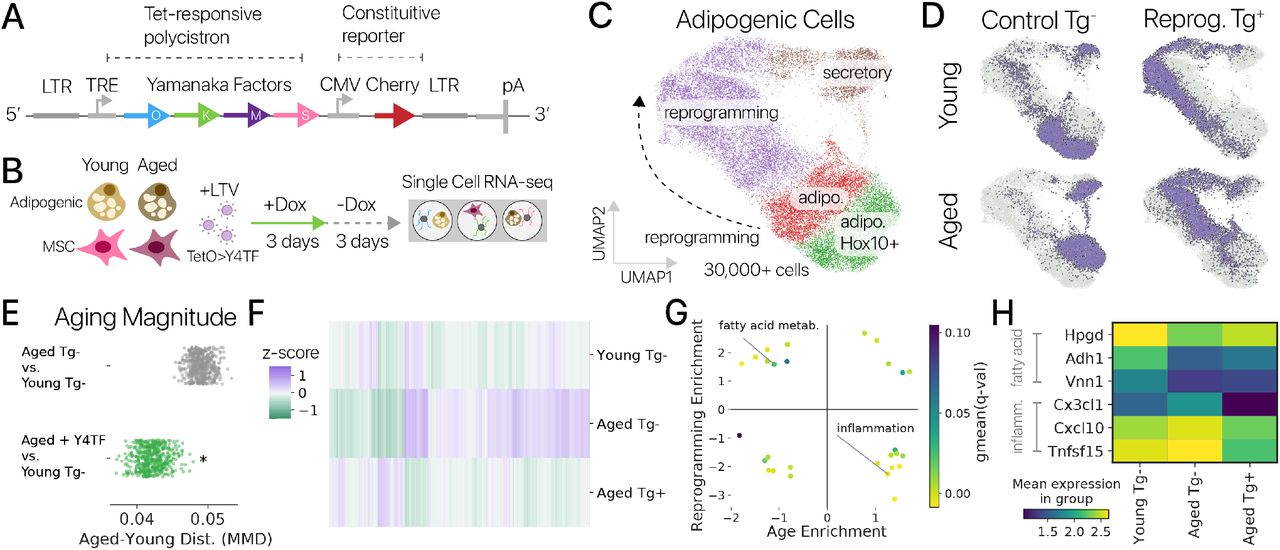
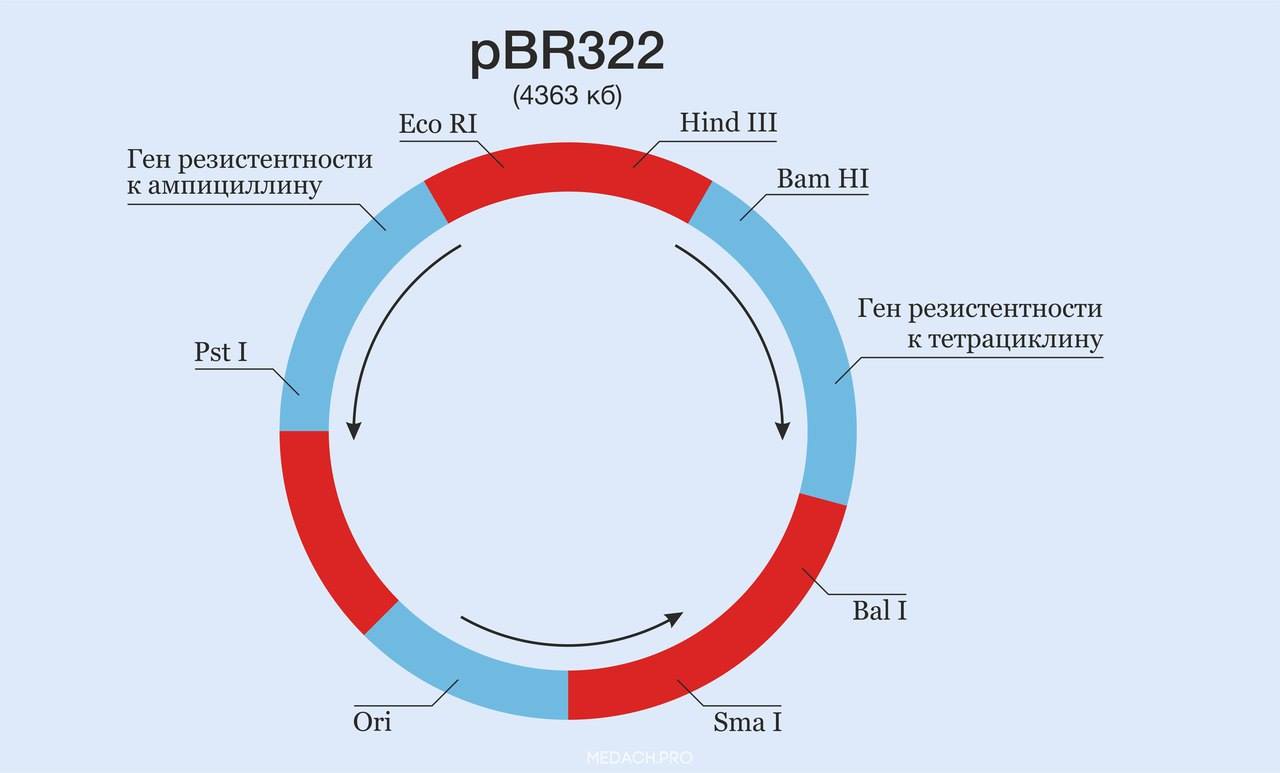 1) Cхема плазмидного вектора pBR322. На плазмидной карте указано расположение некоторых рестриктаз, а также генов антибиотикорезистентности.
1) Cхема плазмидного вектора pBR322. На плазмидной карте указано расположение некоторых рестриктаз, а также генов антибиотикорезистентности.
Клонирование фрагмента ДНК включает в себя определённую последовательность действий: амплификация и очистка клонируемого фрагмента (иначе называется вставка — insert), линеаризация ДНК вектора (такая конформация ДНК более эффективна для получения устойчивых трансфектантов, чем суперспиральная ДНК, что, вероятно, связано со способностью встраиваться в геномную ДНК клетки), связывание (лигирование) вставки и вектора, введение рекомбинантной ДНК в бактериальные клетки (трансформация), отбор и выращивание трансформированных бактерий (селекция), выделение рекомбинантной ДНК из бактерий и перепроверка клонирования. Для каждого этапа уже разработаны, насколько возможно, подробные протоколы и стандарты. Кроме того, наличие широкого спектра реагентов, векторов и штаммов бактерий облегчает работу начинающим исследователям. Теперь всё зависит от выбора стратегии хода эксперимента и правильного подбора всех компонентов.
Выбор вектора зависит от длины клонируемого участка ДНК (неважно, ДНК-копия это или же часть геномной ДНК клеток организма-донора), поскольку векторы отличаются по своей вместительности. К примеру, наиболее часто используемые векторы — производные бактериальных плазмид — способны принимать гетерологичную ДНК длиной от нескольких пар оснований до 6‒8 килобаз (кб) (более длинные вставки будут препятствовать замыканию кольца). Для переноса генов, несколько превышающих размером 10 килобаз, часто применяют фаговые векторы, получаемые из лямбда-фагов, чей геном составляет около 49 килобаз (примерно 40% от данной длины могут быть заменены на чужеродную ДНК). Для ещё более протяжённых фрагментов ДНК (неизменённых частей геномной ДНК, например) в распоряжении учёных есть весьма ёмкие космиды — векторы, включающие в себя cos-участок генома бактериофага лямбда (обеспечивает упаковку ДНК в голову фага), а также имеющие участки, позволяющие реплицироваться по принципу, сходному с тем, что характерен для плазмид. Кроме того, среди векторных систем, способных интегрировать крупные вставки, выделяют искусственные хромосомы, которые могут содержать в себе как гены фагов, так и дрожжей или бактерий, например: PAC (P1 derived artificial chromosomes), BAC (bacterial artificial chromosomes), YAC (yeast artificial chromosomes), MAC (mammalian artificial chromosomes).
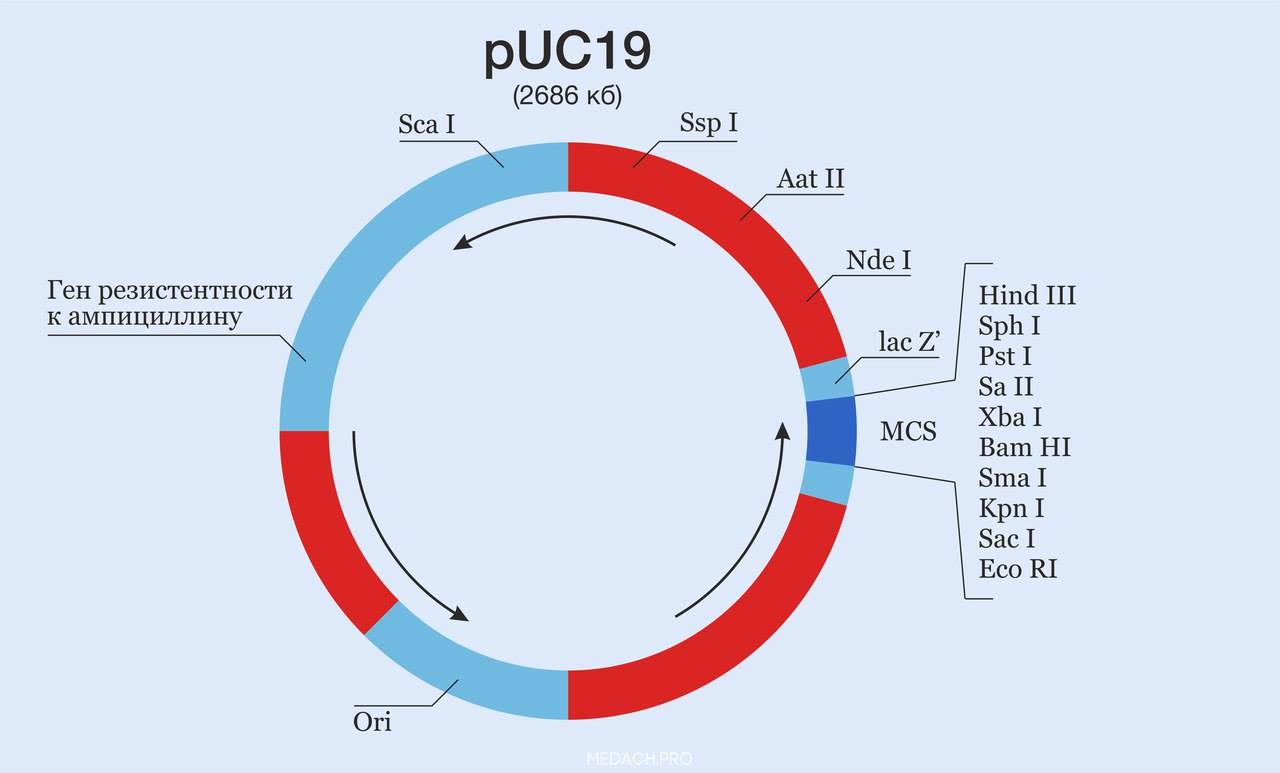 Схема плазмидного вектора pUC19 (UC - потому что разработан в University of California). Указано место начала репликации (ori), ген устойчивости к ампициллину. Рестриктазы, в отличие от pBR322, собраны в одном участке - MCS. Кроме того, полилинкер расположен в пределах последовательности гена lacZ’ (используется для быстрого отбора колоний рекомбинантных бактерий после трансфекции; бело-голубой скрининг). Данный ген уничтожается, как только вектор раскрывается в этом месте рестриктазами, чтобы внести желаемую вставку на его место.
Схема плазмидного вектора pUC19 (UC - потому что разработан в University of California). Указано место начала репликации (ori), ген устойчивости к ампициллину. Рестриктазы, в отличие от pBR322, собраны в одном участке - MCS. Кроме того, полилинкер расположен в пределах последовательности гена lacZ’ (используется для быстрого отбора колоний рекомбинантных бактерий после трансфекции; бело-голубой скрининг). Данный ген уничтожается, как только вектор раскрывается в этом месте рестриктазами, чтобы внести желаемую вставку на его место.
Остановимся более подробно на фагах, чтобы принцип их использования как средства для клонирования и переноса генетического материала стал действительно ясным. Выбор бактериофагов в качестве векторных «доставщиков» ДНК в клетку был связан с тем, что фаги обладают выверенными четырьмя миллиардами лет эволюции естественными механизмами внесения генетического материала в клетку и обеспечивают его размножение клеткой-хозяином, задействуя внутриклеточные механизмы и органоиды. Фаги могут следовать литическому или лизогенному циклу, соответственно разрушая бактериальную клетку после синтеза и выхода из клетки новых фаговых частиц, либо сохраняя свою нуклеиновую кислоту интегрированной в геном бактерии, реплицируясь вместе с ним, что может продолжаться множество генераций, а под влиянием каких-либо внешних условий (например, УФ-излучение) фаговая ДНК может быть снова вырезана. Фаги лямбда имеют довольно характерную структуру, отличающую их, скажем, от филаментных фагов (как М13), и состоят из головы — капсида, внутри которого заключена двухцепочечная молекула ДНК, и хвоста, с помощью которого происходит закрепление бактериального вируса на клетке-мишени для внедрения своей ДНК. Почему же вдруг учёные решили, что можно запросто заменить часть собственного генома такого фага? Было определено, что около трети генома не задействовано в репликации фаговой ДНК. Из этих соображений была разработана структура лямбда-векторов, которая представляет собой конструкцию из трёх больших частей: правое и левое плечи линейной лямбда-ДНК, которые содержат всю необходимую информацию для размножения фага (гены белков для построения головы и хвоста, а также для обеспечения встраивания, репликации и лизиса клетки бактерии). А концы этих плечей ДНК обозначаются как cos (cohesive end sites), каждый из которых, по сути, является цепочкой ДНК в 12 пар оснований. Между плечами располагается та часть фаговой ДНК, которая будет вырезана за счёт того, что имеет по бокам полилинкерные последовательности (содержат сайты рестрикции, узнаваемые рестриктазами; MCS – multiple cloning site), а на её место помещается вставка. Чтобы фаг беспрепятственно попал в клетку и вектор был размножен, требуется следить за размером встраиваемой ДНК. Для корректной упаковки вирусной частицы длина ДНК должна в среднем составлять до 50 кб. Правое и левое плечо вместе уже составляют около 30 кб в длину, поэтому встраиваемый участок должен быть размером в 10‒20 кб. Итак, если инфекционные вирусные частицы правильно сформированы, после их упаковки следует инфицирование бактерий, которые предварительно высевают на агар. Через какое-то время в тех местах, где бактериальные клетки подверглись лизису в результате фаговой активности, образуются легко различимые пятна, где можно обнаружить высвобождённые фаги, из которых и выделяется рекомбинантная ДНК.
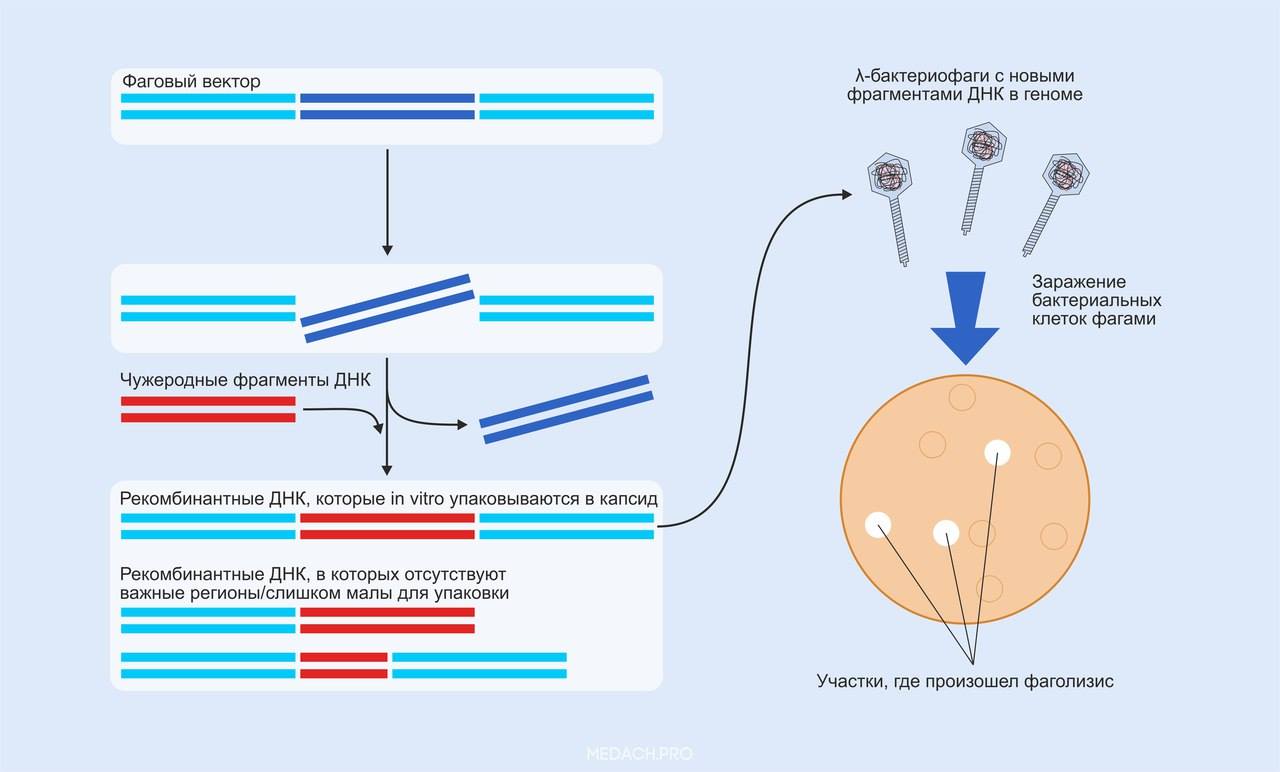 Схема проведения клонирования ДНК с помощью бактериофагов. Происходит вырезание фрагмента исходной вирусной ДНК рестриктазами; чужеродные фрагменты связываются с оставшимися участками и формируются рекомбинантные ДНК, которые in vitro упаковываются в капсид фага. После инфицирования и лизиса бактериальных клеток можно выделять рекомбинантную ДНК.
Схема проведения клонирования ДНК с помощью бактериофагов. Происходит вырезание фрагмента исходной вирусной ДНК рестриктазами; чужеродные фрагменты связываются с оставшимися участками и формируются рекомбинантные ДНК, которые in vitro упаковываются в капсид фага. После инфицирования и лизиса бактериальных клеток можно выделять рекомбинантную ДНК.
3) Схема проведения клонирования ДНК с помощью бактериофагов. Происходит вырезание фрагмента исходной вирусной ДНК рестриктазами; чужеродные фрагменты связываются с оставшимися участками и формируются рекомбинантные ДНК, которые in vitro упаковываются в капсид фага. После инфицирования и лизиса бактериальных клеток можно выделять рекомбинантную ДНК.
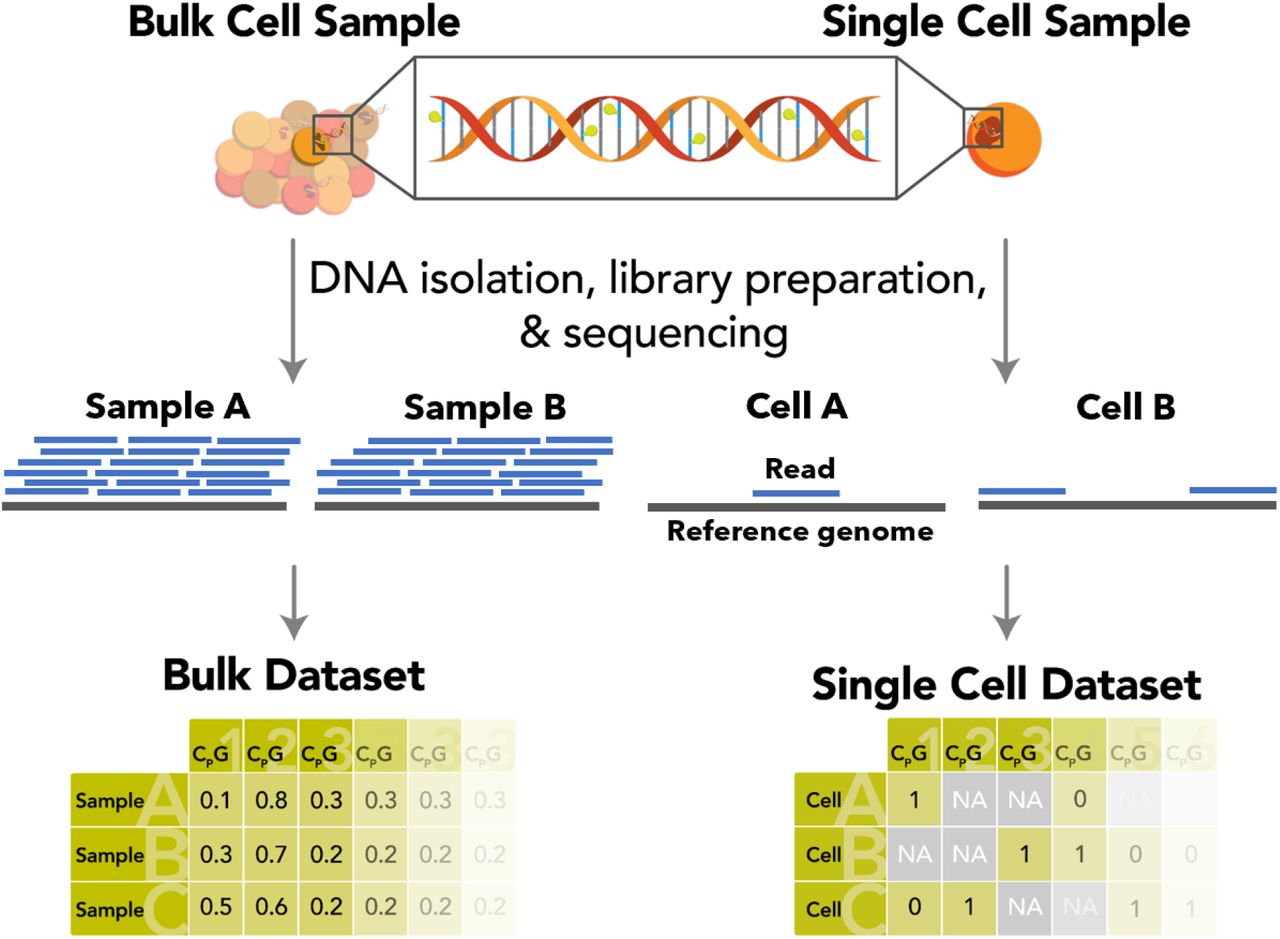
Итак, рассмотрев кратко возможности клонирования рекомбинантной ДНК, обратимся к её доставке в целевые клетки. Как уже упоминалось выше, методика подбирается в зависимости от типа принимающих клеток. Обычно используемые культуры клеток млекопитающих довольно хорошо поддаются трансфекции (невирусная доставка ДНК), но в ряде случаев приходится прибегать к трансдукции гетерологичной ДНК при помощи рекомбинантных вирусов.
Вирусные векторные системы, являясь альтернативой трансфекции, располагают несколькими вариантами векторов, среди которых аденовирусные, ретровирусные, лентивирусные, Semliki-Forest, вирус Синдбис. Разные типы вирусов обладают своими плюсами и минусами (поэтому, повторюсь, их подбирают сообразно целям трансдукции). К примеру, рекомбинантные аденовирусные системы от штамма Ad5 довольно эффективно инфицируют сразу большое количество клеток млекопитающих различных типов независимо от степени способности клеток к делению (хотя наиболее годятся для неделящихся клеток). Привлекательной чертой данных векторов представляется возможность контроля степени экспрессии внедряемого гена, поскольку этот аспект зависит от соотношения количества вирусных частиц к числу клеток (то есть, отрегулировав концентрацию вирусных частиц в суспензии, можно повлиять на уровень экспрессии гена). Эта особенность позволяет, кроме прочего, инфицировать клетки несколькими рекомбинантными аденовирусами, обеспечив экспрессию нескольких необходимых белков одновременно. Однако внесённая таким образом ДНК располагается в клетках эписомно, а значит, это большой минус для интенсивно делящихся клеток, поскольку по завершению клеточного цикла информация об экспрессии гетерологичного гена утрачивается дочерними клетками. Немаловажной особенностью вирусных векторов (в том числе и аденовирусных) является сохранность способности вирусных частиц взаимодействовать с рецепторами клеток-мишеней (например, FGFR1) посредством специфических лигандов, расположенных на поверхности вектора, что обуславливает осуществление клатрин-зависимого эндоцитоза. Разнообразные типы и серотипы встречающихся вирусов имеют множество белков вирусных оболочек, что даёт широкий спектр лигандов и лежит в основе тканеспецифичности таких вирусных векторов. А создание рекомбинантных серотипов с определёнными поверхностными белками обуславливает трансдукцию только специфических типов клеток, а также может оказывать влияние на выбор способа введения. Специфичность векторов относительно клеток в пределах одной ткани настраивается также с помощью промоторов векторов (например, RK-промотор (родопсинкиназный) для фоторецепторов сетчатки).
Напоследок захватим и невирусные способы транспорта генов в клетки. Например, на вооружении исследователей имеются такие физические методы, как электропорация, когда подача коротких электрических импульсов делает клеточную стенку на некоторое время проницаемой для вносимой ДНК, которая никак не упакована. Другим способом является формирование комплекса терапевтического гена с наночастицами, который захватывается и поглощается клетками путем рецептор-опосредованного эндоцитоза. Также внесение гетерологичной ДНК возможно путем трансфекции. При трансфекции ДНК, как правило, вносится в форме преципитата, комплекса с полимерами или упакованная в липидные везикулы и активно поглощается клетками. В ходе липофекции in vitro клетки инкубируют после высевания до достижения ими довольно высокого уровня конфлюэнтности; для трансфекции подготавливаются комплексы ДНК с катионными липидами. Затем клетки отмываются, и на них наносится подготовленный раствор с комплексами ДНК-липиды, после чего клетки инкубируют несколько часов. По окончании трансфекционный раствор удаляется, и клетки снова высеваются на среду, через некоторое время клетки можно исследовать на предмет активности гена.
На этом наш краткий обзор завершается. Важной его целью было показать то, насколько непросто внести чужеродную ДНК в клетки бактерий или многоклеточного организма, как много подводных камней ждёт на пути, несмотря на обширность имеющихся материалов для создания рекомбинантных ДНК. Участок ДНК нелегко поместить в клетку, и довольно сложно сохранить его в последующих генерациях, тем не менее, методы генной инженерии весьма перспективны относительно генной терапии множества заболеваний, что мы обязательно обсудим в последующих постах.
Автор статьи: Yael Demedetskaya
Источники:
M. Wink Molekulare biotechnologie, 2. Auflage, Wiley-VCH verlag, 2011
Фрешни Р.Я. Культура животных клеток, Бином, 2010
И.Ф. Жимулев Общая и молекулярная генетика, Сиб. унив. изд-во,2007
D. Ganten, K. Ruckpaul Grundlagen der molekularen Medizin, 2. Auflage, Springer, 2003
Подробнее по теме: ГЕННАЯ ТЕРАПИЯ
 Астроциты — это клетки, которые в норме должны защищать нейроны и помогать им образовывать новые связи. Однако в некоторых случаях астроциты оказываются агрессивны по отношению к нейронам и другим клеткам нервной системы.
Астроциты — это клетки, которые в норме должны защищать нейроны и помогать им образовывать новые связи. Однако в некоторых случаях астроциты оказываются агрессивны по отношению к нейронам и другим клеткам нервной системы.